

Foundation B (Y2)
Question 1:
Answer: D) Increased expression of N-cadherin and loss of E-cadherin
Explanation: EMT is a process where epithelial cells (which are usually organized and have an apical and basal side) transform into mesenchymal cells (which are more mobile and invasive). This transition is crucial for cancer metastasis because it allows cancer cells to detach from the primary tumour and invade other tissues. E-cadherin is a cell adhesion molecule that keeps epithelial cells tightly connected. During EMT, E-cadherin is downregulated, causing the loss of cell-to-cell adhesion. N-cadherin is typically expressed in mesenchymal cells and promotes mobility. Its upregulation helps cancer cells move and invade surrounding tissues. Matrix metalloproteinases (MMPs) are also upregulated in EMT. They help break down the extracellular matrix (ECM), which cancer cells need to pass through to invade new tissues.
A) is incorrect as this is the opposite of what happens during EMT. E-cadherin is lost, and N-cadherin is gained, not the other way around.
B) is incorrect as EMT involves increased MMPs to help cancer cells break through tissue barriers. A decrease would hinder invasion.
C) is incorrect as transition from motile mesenchymal cells to stationary epithelial cells describes the reverse process, called Mesenchymal-to-Epithelial Transition (MET). In EMT, cells become more mobile.
E) is incorrect as tumour-suppressor genes (like p53) are often inactivated during cancer progression. EMT is associated with oncogene activation, not tumour suppression.
Question 2:
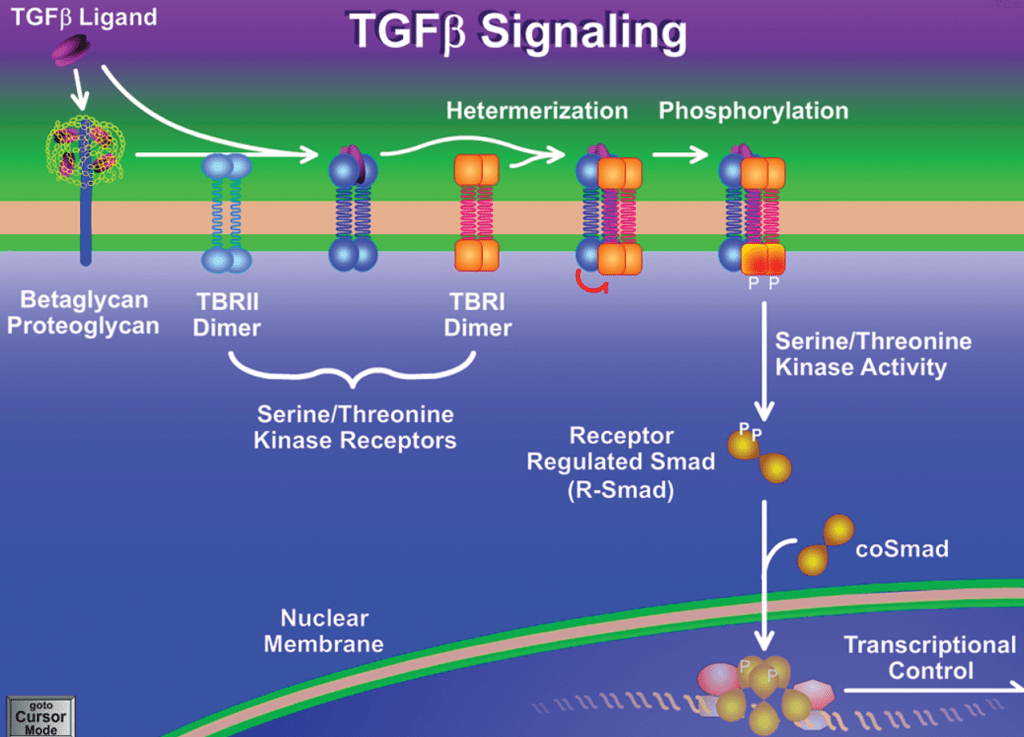
Answer: B) Loss of TGF-β RII function leads to uncontrolled cell proliferation.
Explanation: TGF-β (Transforming Growth Factor-beta) is a cytokine that regulates cell growth, differentiation, and apoptosis. In normal cells, TGF-β signalling helps control cell proliferation and suppress tumours. It activates SMAD proteins, which enter the nucleus and inhibit growth-promoting genes. In colorectal cancer (CRC), mutations in TGF-β RII or SMAD4 lead to loss of tumour-suppressive function. When TGF-β signalling is disrupted, cells lose growth control, contributing to cancer progression.
A) is incorrect as although TGF-β signalling promotes tumour suppression by inhibiting cell proliferation is true in normal cells. In cancer, mutations can disrupt this pathway. Loss of TGF-β RII function leads to uncontrolled growth.
C) is incorrect as SMAD4 mutations inactivate TGF-β signalling, reducing its tumour-suppressive effects.
D) is incorrect as TGF-β signalling is highly relevant. Mutations in this pathway are a common feature of CRC/bowel cancer.
E) is incorrect as TGF-β RII activates SMAD proteins, which then suppress cell growth not the opposite.
Question 3:
Answer: E) Initiates DNA repair or apoptosis in response to cellular stress
Explanation: P53 is a tumour suppressor protein often called the “guardian of the genome” because it helps maintain genomic stability. When cells experience stress (like DNA damage), p53 is activated and does one of two things: DNA repair: Pauses the cell cycle to allow repair mechanisms to fix damaged DNA and Apoptosis: If damage is irreparable, p53 triggers programmed cell death to prevent the proliferation of abnormal cells. Mutations in the TP53 gene (which codes for p53) are found in over 50% of cancers, leading to uncontrolled cell growth.
A) is incorrect as oncogenes are what promote cell growth and are typically activated in cancer. p53, as a tumour suppressor, does the opposite—it inhibits abnormal cell proliferation.
B) is incorrect as telomerase activation is a feature of cancer cells, allowing them to avoid aging. P53 inhibits this process by promoting senescence (cell aging) in damaged cells.
C) is incorrect as p53 does not regulate the immune system directly. It focuses on cell cycle control and apoptosis.
D) is incorrect as angiogenesis (formation of new blood vessels) is promoted by factors like VEGF in tumours, p53 helps inhibit angiogenesis.
Question 4:
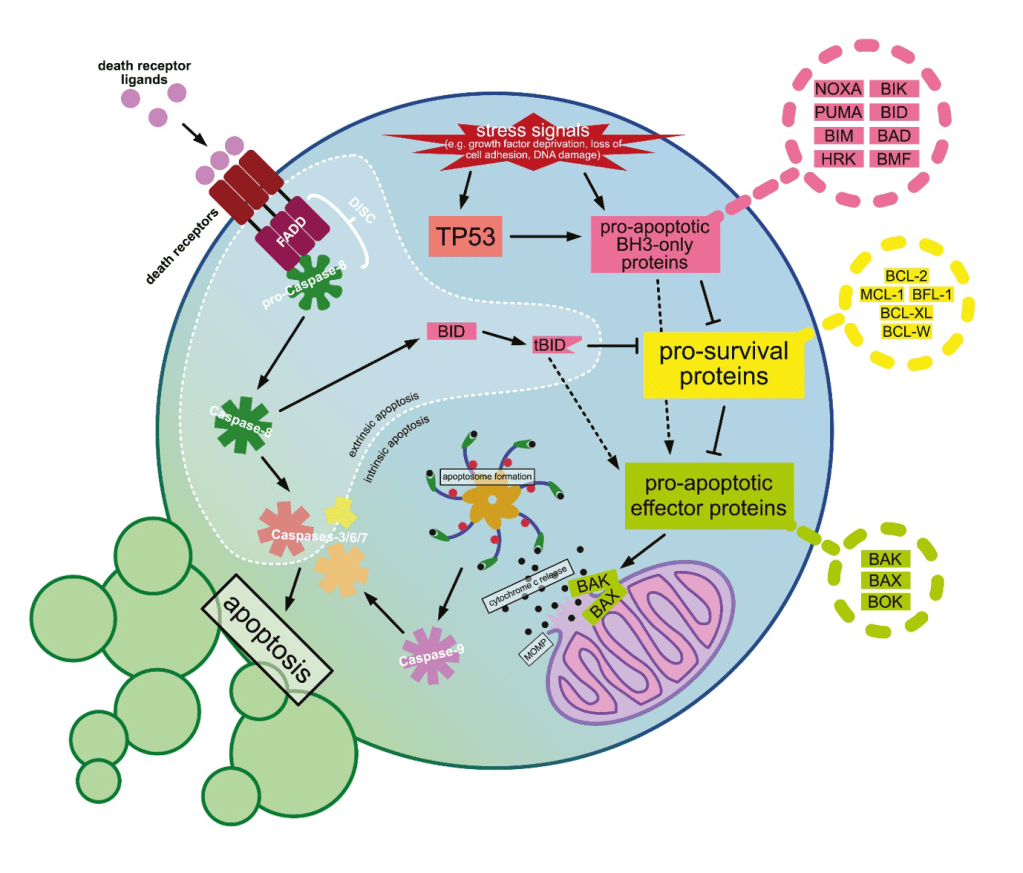
Answer: C) Loss of p53 function
Explanation: p53 is crucial for triggering apoptosis when cells experience DNA damage. Loss or mutation of p53 prevents apoptosis, allowing damaged cells to survive and proliferate, which is a common mechanism in many cancers.
A) is incorrect as proteins like Bax and Bak are proteins that promote apoptosis by forming pores in the mitochondrial membrane.
B) is incorrect as proteins like Bcl-2 are anti apoptotic so reducing Bcl-2 would promote apoptosis. Cancer cells often overexpress Bcl-2 to avoid cell death, they do not downregulate it.
D) is incorrect as caspases are key executioners of apoptosis. Increased activation would lead to cell death, not evasion.
E) is incorrect as death receptors (like Fas) initiate apoptosis; cancer cells often downregulate these to survive.
Question 5:
Answer: B) Promoting p53 degradation
Explanation: MDM2 is an E3 ubiquitin ligase that binds to p53, tagging it for proteasomal degradation. Under normal conditions, MDM2 keeps p53 levels low to prevent unnecessary apoptosis. However, when DNA damage occurs, p53 is phosphorylated, preventing MDM2 binding, allowing p53 to accumulate and activate cell-cycle arrest or apoptosis.
A) is incorrect as MDM2 inhibits p53’s activity, not enhances it.
C) is incorrect as MDM2 does not influence p53 production; it controls its degradation.
D) is incorrect as phosphorylation blocks MDM2 binding, allowing p53 activation.
E) is incorrect as MDM2’s role is to suppress p53’s apoptotic function, not activate it.
Question 6:
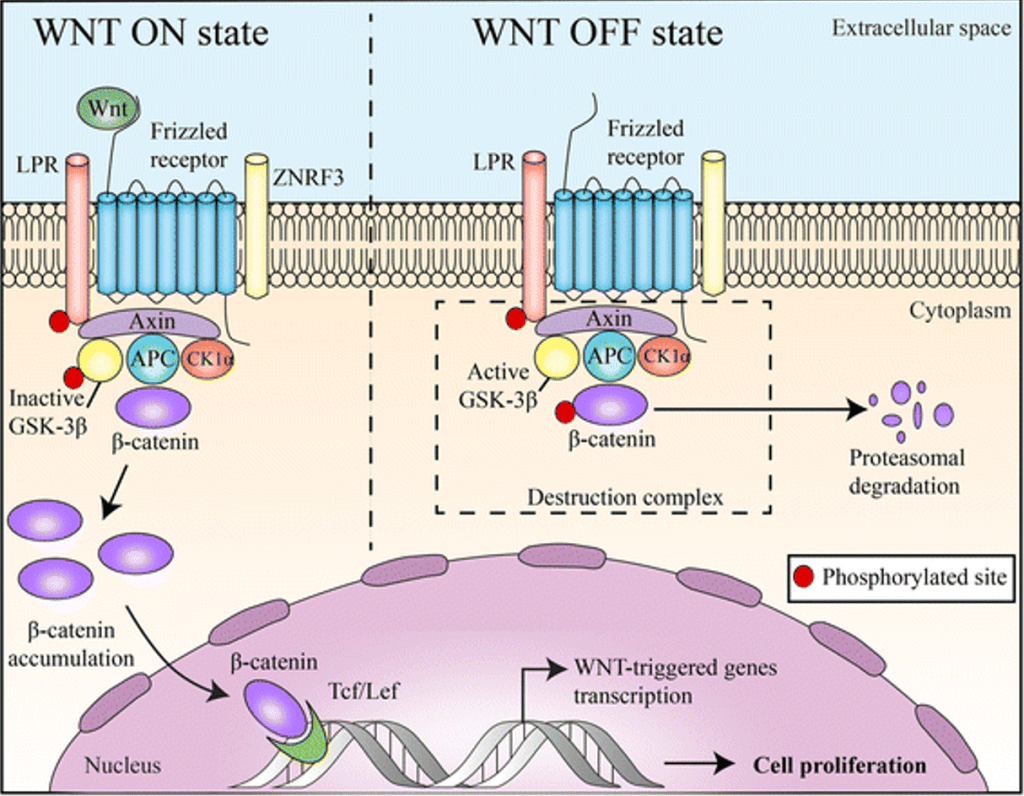
Answer: A) Accumulates and enters the nucleus
Explanation: Wnt proteins (ligands) are signalling molecules that initiate the pathway. When Wnt binds to its receptor (Frizzled) on the cell membrane, it forms a complex with another co-receptor called LRP (Lipoprotein Receptor-related Protein). The Wnt-Frizzled-LRP complex activates a protein called Dishevelled (Dvl) inside the cell. Dvl becomes phosphorylated and inhibits the destruction complex that normally degrades β-catenin. Under normal conditions (without Wnt signalling), the destruction complex consists of: APC (Adenomatous Polyposis Coli), Axin, GSK-3β (Glycogen Synthase Kinase 3-beta) and CK1 (Casein Kinase 1). This complex phosphorylates β-catenin, marking it for ubiquitination and subsequent proteasomal degradation. When Wnt signalling is active, Dishevelled (Dvl) inhibits GSK-3β and Axin, preventing the destruction complex from functioning. This stops β-catenin from being degraded. Instead, it accumulates in the cytoplasm and then translocates into the nucleus. In the nucleus, β-catenin binds to TCF/LEF (T-cell factor/lymphoid enhancer-binding factor) transcription factors. This activates genes involved in cell proliferation, survival, and differentiation. The target genes activated by β-catenin promote cell division and growth, which is crucial in processes like tissue regeneration. However, dysregulation of this pathway (e.g., mutations in APC or β-catenin) can lead to uncontrolled cell proliferation and cancer, like colorectal cancer.
B) is incorrect as Beta catenin is degraded by the proteasome without Wnt signalling.
C) is incorrect as the destruction complex is inhibited in the presence of Wnt, it doesn’t remain bound.
D) is incorrect and by binding to frizzled prevents the destruction of beta catenin, therefore allowing it to enter the nucleus.
E) is incorrect as beta catenin stimulates cell proliferation, it does not inhibit it.
Question 7:
Answer: C) Multiple sub-clones with distinct mutations arise from a common ancestor
Explanation: Branched evolution occurs when multiple sub-populations (clones) of cancer cells diverge from a single ancestor, each acquiring unique mutations. This leads to genetic heterogeneity within a tumour, making treatment challenging.
A) is incorrect as sequential mutations produce one dominant clone describes linear evolution, not branched.
B) is incorrect as heterogeneity is key in branched evolution.
D) is incorrect as some cancer cells evolve linearly without variation but not all.
E) is incorrect as branching involves multiple mutations in different clones.
Question 8:
Answer: E) A tumour containing tissues derived from all three germ layers (ectoderm, mesoderm, endoderm).
Explanation: Teratomas are tumours that contain tissues from all three germ layers, they are most found in the gonads (ovaries or testes).
Ectoderm: May form skin or neural tissue.
Mesoderm: Can develop into bone, muscle, or cartilage.
Endoderm: Can produce glandular tissues like gastrointestinal lining, or epithelium that lines organs.
A) is incorrect as tumours from mesenchymal tissue are called sarcomas.
B) is incorrect as tumours from a single germ layer are not teratomas; they are usually more specialized and called mixed tumours.
C) is incorrect because a malignant tumour composed of epithelial cells is a carcinoma.
D) is incorrect because a benign tumour containing adipose tissue is called a lipoma.
Question 9:
Answer: D) The formation of dense, fibrous tissue around a tumour.
Explanation: Desmoplasia refers to the growth of dense, fibrous connective tissue around a tumour, primarily due to the increased production of collagen and other extracellular matrix components. This process is driven by the interaction between tumour cells and stromal fibroblasts mainly, leading to a hard, firm texture characteristic of many carcinomas. This is usually common in carcinomas (malignant epithelial tumours) and represents a reaction by the body’s stromal cells to the invasive cancer cells.
A) is incorrect as the growth of abnormal blood vessels is called angiogenesis.
C) is incorrect as the spread of tumour cells to distant organs is known as metastasis.
B) is incorrect because necrosis refers to cell death within a tumour, often due to poor blood supply.
E) is incorrect because uncontrolled proliferation of epithelial cells describes carcinogenesis but not desmoplasia.
Question 10:
Answer: A) Pleomorphism refers to variations in size and shape of cells, while anaplasia indicates a lack of differentiation.
Explanation: Pleomorphism is the variation in size and shape of cells and their nuclei within a tumour, this is common in malignant tumours and reflects abnormal differentiation. Anaplasia is a lack of differentiation, meaning the cells lose their normal structure and function. Anaplastic cells exhibit high-grade malignancy, pleomorphism, hyperchromatic nuclei, and abnormal mitosis.
Question 11:
Answer: B) It indicates the proportion of tumour cells actively dividing.
Explanation: The growth fraction refers to the proportion of tumour cells that are actively undergoing division (in the cell cycle). High growth fractions are seen in rapidly growing tumours and indicate a higher malignancy potential, such as lymphoma and leukaemia. However, tumours with a high growth fraction are often more responsive to chemotherapy, since it targets dividing cells in the cell cycle.
A) is incorrect because the number of mutations does not determine the growth fraction.
C) is incorrect as this describes angiogenesis, not growth fraction.
D) is incorrect as metastatic potential refers to the ability of a tumour to spread, not the proportion of dividing cells.
E) is incorrect because the tissue origin is determined by histological examination.
Question 12:
Answer: C) Stabilise and mature newly formed blood vessels.
Explanation: After endothelial cells form new blood vessels during angiogenesis, pericytes are recruited to wrap around the endothelial cells. They help stabilise the newly formed blood vessels, preventing them from being too leaky or fragile. This is critical because unstable vessels can lead to inefficient oxygen and nutrient delivery. Pericytes also regulate endothelial cell proliferation, migration, and differentiation during angiogenesis by secreting pro-angiogenic factors, like VEGF (vascular endothelial growth factor), which stimulates endothelial cells to grow and migrate. Conversely, they also produce anti-angiogenic factors to help terminate vessel growth when sufficient blood vessels have been formed. This balance is crucial for normal angiogenesis.
A) is incorrect as although pericytes influence endothelial cell survival, their primary role is not preventing apoptosis.
B) is incorrect as pericytes do not affect immune cell proliferation, this is the function of cytokines and chemokines.
D) is incorrect as pericytes decrease permeability to stabilize blood vessels. In cancer, tumour-associated pericytes are often fewer or improperly recruited, which contributes to the instability of the tumour vasculature, this makes blood vessels leaky, aiding in tumour metastasis by allowing cancer cells to enter the bloodstream more easily.
E) is incorrect as pericytes do not replace endothelial cells but support and interact with them.
Question 13:
Answer: D) The TME promotes immune suppression by recruiting T-regulatory cells and polarizing macrophages to an M2 phenotype.
Explanation: The TME is composed of stromal cells, immune cells, ECM, and blood vessels that interact with cancer cells to promote their survival and progression. The TME actively recruits T-regulatory cells (Tregs) via chemokines and polarizes macrophages into the M2 macrophages which suppress anti-tumour responses and secrete growth-promoting cytokines. Tregs release immunosuppressive cytokines such as TGF-β and IL-10, directly inhibiting cytotoxic CD8+ T cells and natural killer (NK) cells, reducing the immune system’s ability to attack tumour cells. This immunosuppressive environment allows cancer cells to evade immune detection, promoting tumour growth, invasion, and metastasis.
A) is incorrect as although apoptosis of some immune cells may occur, the primary mechanism of immune evasion is functional suppression of immune cells (e.g., Tregs inhibiting CD8+ T cells).
B) is not the best answer as while CAFs secrete matrix metalloproteinases (MMPs) to degrade the ECM, their role is supportive, not the primary mechanism of TME-driven progression.
C) is incorrect as angiogenesis in tumours is abnormal and chaotic, leading to leaky, poorly functional blood vessels that enhance hypoxia and metastasis.
E) is incorrect as although CD8+ T cells and NK cells are tumour-suppressive, their activity is suppressed within the TME through cytokines (e.g., TGF-β, IL-10) and immune checkpoint molecules (e.g., PD-L1).
Question 14:
Answer: A) TILs are recruited by chemokines secreted by tumour cells but are rendered ineffective by immune checkpoints like PD-1/PD-L1.
Explanation: Tumour infiltrating leukocytes (TIL) are immune cells, such as T cells, B cells, and natural killer (NK) cells, found within the tumour microenvironment (TME). Tumour cells release chemokines (e.g., CXCL9, CXCL10) to recruit immune cells, including TILs, into the TME. Once present, TILs, particularly cytotoxic T cells (CD8+), are inactivated. For example, tumour cells expressing PD-L1 bind PD-1 on T cells, inhibiting their cytotoxic function. In addition, tumours recruit Tregs, which suppress TIL function via IL-10 and TGF-β. This is useful to know clinically as immune checkpoint inhibitors (e.g., anti-PD-1 therapy) aim to restore TIL functionality.
B) is incorrect as TILs recognize tumour neoantigens but are suppressed by the immunosuppressive TME.
C) is incorrect as while Tregs are part of TILs, they are not the only subset; CD8+ and CD4+ effector T cells are also present.
D) is incorrect as VEGF secretion promotes angiogenesis, not TIL recruitment.
E) is incorrect as inflammation can attract immune cells, but TIL recruitment is primarily chemokine mediated.
Question 15:
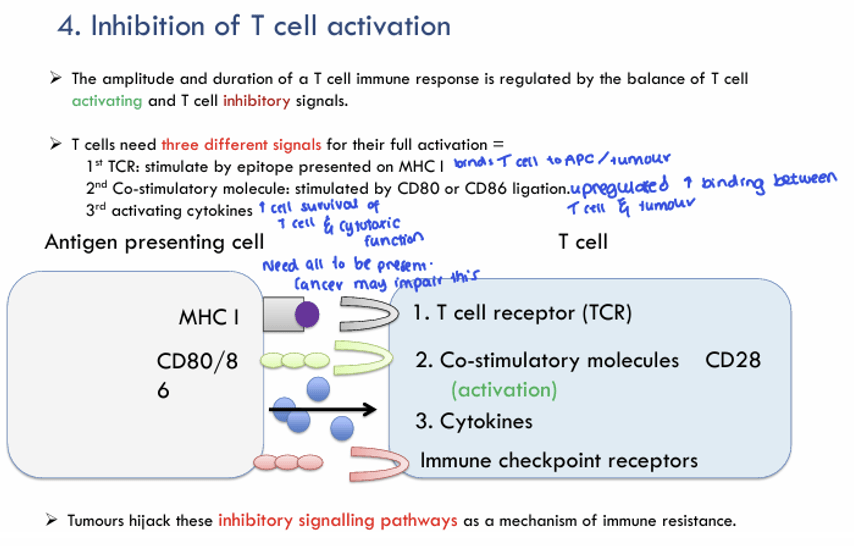
Answer: C) The secretion of IL-10
Explanation: IL-10 is an immunosuppressive cytokine. It does not contribute to T cell activation. Instead, it plays a role in dampening immune responses to prevent overactivation and tissue damage, particularly in chronic infections and autoimmune conditions. IL-10 is secreted by regulatory T cells (Tregs) and other immune cells like macrophages to limit inflammation.
A) is incorrect as TCR binding to an antigen presented on MHC is the first signal required for T cell activation. The T-cell receptor (TCR) binds to the antigen presented on MHC (major histocompatibility complex) molecules on the surface of antigen-presenting cells (APCs). This is the essential first step in initiating an immune response.
B) is incorrect as co-stimulation via CD28 and CD80/86 is the second signal required for full T cell activation. Co-stimulatory molecules (CD80/CD86) on APCs bind to CD28 on T cells, promoting full T cell activation. Without this signal, the T cell may become anergic (unresponsive).
D) is incorrect because cytokine signals, such as IL-2 is a crucial activation cytokine that promotes the proliferation and survival of activated T cells. It is produced following TCR engagement and is necessary for the full activation of T cells.
E) is incorrect because as explained, IL-10 does not contribute to T cell activation, but rather to immune suppression.
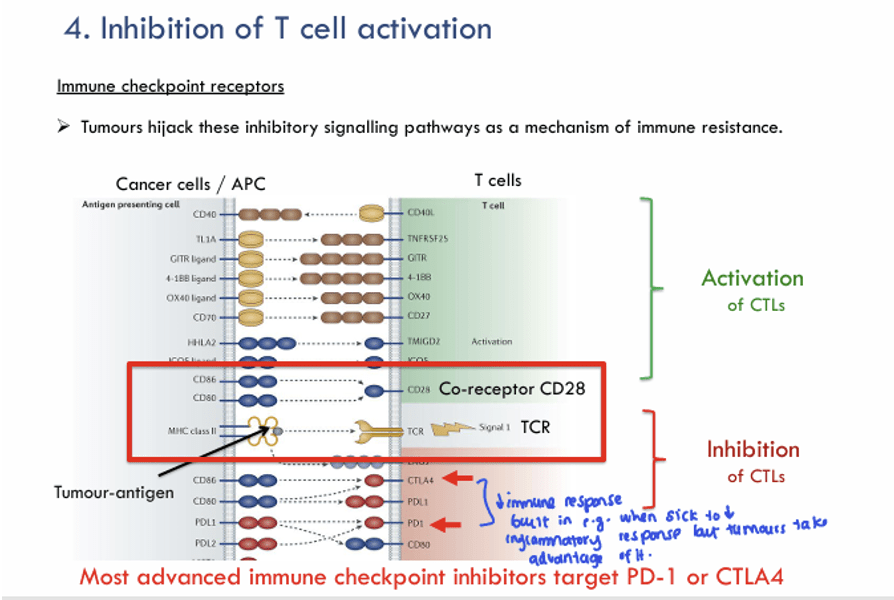
Question 16:
Answer: B) Nivolumab
Explanation: Nivolumab is an anti-PD-1 antibody. PD-1 is an inhibitory receptor on T cells that, when bound to its ligand PD-L1 on tumour cells it suppresses T cell activation. By blocking PD-1, nivolumab prevents this inhibitory signal, thus enhancing T cell function and promoting anti-tumour immune responses.
A) is incorrect as Ipilimumab is an anti-CTLA-4 antibody. It targets CTLA-4, an inhibitory receptor that normally competes with CD28 for co-stimulatory signals on T cells. By blocking CTLA-4, ipilimumab enhances T cell activation and response to cancer. However, it does not target PD-1.
C) is incorrect because Rituximab is an antibody that targets CD20 on B cells, primarily used in treating B cell malignancies like non-Hodgkin lymphoma. It does not target PD-1.
D) is incorrect as Trastuzumab is an antibody targeting HER2/neu, a receptor that is overexpressed in certain breast cancers. It does not interact with PD-1 or immune checkpoint pathways.
E) is incorrect as Cetuximab is an anti-EGFR (epidermal growth factor receptor) antibody used to treat cancers like colorectal and head/neck cancers. It does not block PD-1 or affect immune checkpoints.
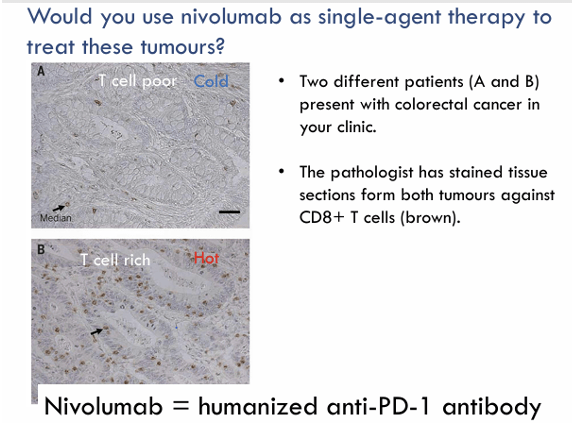
Question 17:
Answer: E) CD80/CD86
Explanation: CD80 and CD86 are co-stimulatory molecules on antigen-presenting cells (APCs) that bind to CD28 on T cells, providing the second signal necessary for full T cell activation. This co-stimulation is essential for T cell survival and function.
A) is incorrect because CTLA-4 is an inhibitory receptor on T cells that competes with CD28 for binding to CD80 and CD86. It negatively regulates T cell activation by delivering inhibitory signals after T cell activation.
B) is incorrect as PD-1 is another inhibitory receptor on T cells. It interacts with PD-L1 (on tumour cells) to inhibit T cell function. It is not involved in co-stimulation but rather in immune suppression.
C) is incorrect because TGF-β is an inhibitory cytokine that plays a key role in immune suppression. It can promote regulatory T cell differentiation and inhibit the activation of effector T cells.
D) is incorrect because IL-10 is an immunosuppressive cytokine, not a co-stimulatory molecule. It plays a role in suppressing immune responses and is produced by regulatory T cells (Tregs) and other immune cells such as Myeloid Derived Suppressor Cells.

Question 18:
Answer: A) Suppressing T cell activity
Explanation: MDSCs consist of immature myeloid cells that can differentiate into macrophages, dendritic cells, or granulocytes, but in cancer, they remain in an immature and suppressive state. They are not fully differentiated and are thought to be recruited to tumours to inhibit immune responses, which helps the tumour evade immune surveillance. They secrete various immunosuppressive cytokines and enzymes that contribute to the suppression of T cell function such as:
Arginase which is an enzyme that depletes arginine in the microenvironment. Arginine is essential for the activation and proliferation of T cells, so its depletion effectively inhibits T cell responses. Arginase also induces T cell apoptosis.
Nitric Oxide (NO), a molecule that downregulates T cell receptor (TCR) signalling and induces T cell apoptosis.
TGF-β (Transforming Growth Factor-beta): an immunosuppressive cytokine that inhibits the differentiation and function of cytotoxic T cells (CTLs). It also promotes the generation of regulatory T cells (Tregs), further increasing immune suppression.
CCL2, CCL3 and CCL4 are chemokines are involved in recruiting more immune suppressive cells to the tumour site, helping to amplify the suppression of immune responses.
Question 19:
Answer: D) TGF-β
Explanation: TGF-β is a key immunosuppressive cytokine produced by regulatory T cells (Tregs). It inhibits the activation and proliferation of effector T cells, including cytotoxic T cells and helper T cells. TGF-β also promotes the secretion of other suppressive cytokines, such as IL-10 and IL-35, that further dampen immune responses and promote immune tolerance.
A) is incorrect as IL-2 is primarily secreted by activated T cells, particularly CD4+ helper T cells, and is essential for T cell proliferation. It is not a suppressive cytokine secreted by Tregs.
B) is incorrect as IFN-γ is secreted by Th1 cells and is important for enhancing immune responses, particularly in the context of infections and cancer. It is pro-inflammatory, not immunosuppressive like TGF-β.
C) is incorrect as TNF-α is a pro-inflammatory cytokine produced by activated immune cells like macrophages and T cells. It plays a role in inflammation, apoptosis, and fighting infections, but is not involved in immune suppression.
E) is incorrect as IL-12 is produced by dendritic cells and macrophages and plays a key role in promoting the Th1 immune response. It is not secreted by Tregs and is associated with immune activation, not suppression.
Question 20:
Answer: B) non-homologous end-joining (NHEJ)
Explanation: NHEJ repairs double-strand breaks without a template, often leading to small insertions or deletions at the repair site, making it inherently mutagenic.
A) is incorrect because homologous recombination is a high-fidelity repair mechanism, it uses a sister chromatid as a template, which minimises errors.
C) is incorrect because base excision repair involves precise repair of single-base damage using a DNA glycosylase to remove damaged bases, followed by high-fidelity polymerases such as Pol B.
D) is incorrect because mismatch repair corrects replication errors during the s phase of the cell cycle, which maintains genomic integrity rather than introducing mutations.
E) is incorrect because nucleotide excision repair (NER) removes bulky DNA adducts or UV-induced damage (e.g., thymine dimers) in the G1 phase of the cell cycle with high fidelity, making it non-mutagenic.
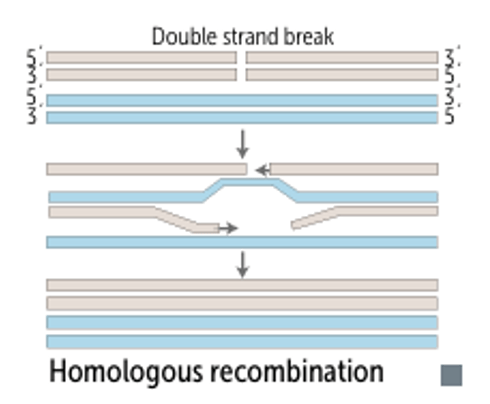

Question 21:
Answer: E) Requires Cockayne Syndrome proteins CSA and CSB
Explanation: Transcription-coupled NER is specialized for repairing lesions that block transcription. CSA and CSB recognize stalled RNA polymerase at the site of damage and recruit the NER machinery.
A) is incorrect because XPC is involved in global genome NER, not transcription-coupled NER.
B) is incorrect because homologous recombination is used for double-strand break repair, not NER.
C) is incorrect because interstrand crosslink repair involves multiple pathways, including homologous recombination and Fanconi anaemia proteins, not NER.
D) is incorrect because oxidative damage is repaired by base excision repair (BER), not NER.
Question 22:
Answer: A) Detects single strand breaks (SSBs) and recruits repair machinery
Explanation: Upon binding to SSBs, PARP1 synthesizes poly(ADP-ribose) chains to signal the damage and recruit repair proteins such as XRCC1 and DNA polymerase β (Pol β). This process is a key component of the Base Excision Repair (BER) pathway.
B) is incorrect because double-strand breaks (DSBs) are repaired by pathways like homologous recombination (HR) and non-homologous end-joining (NHEJ). And the proteins involved in HR are Rad51 and BRCA1/2 , not PARP1.
C) is incorrect because mismatch repair (MMR) involves proteins like MSH2, MSH6, MLH1, and PMS2, which correct mismatched bases that escape proofreading during replication. PARP1 does not participate in mismatch repair.
D) is incorrect because PARP1 is not an exonuclease. It is a signalling protein that detects SSBs and facilitates repair but does not directly degrade DNA.
E) is incorrect because although there are helicases like TFIIH which are involved in transcription-coupled repair and nucleotide excision repair (NER), PARP1 is not directly involved in these processes as it is part of base excision repair (BER) not NER.
Question 23:
Answer: D) Induction of chronic inflammation and oxidative stress
Explanation: H. pylori colonise the stomach leading to chronic gastritis, which is characterised by infiltration of immune cells like neutrophils and macrophages. These immune cells release reactive oxygen species (ROS) and reactive nitrogen species (RNS), which can cause DNA damage, mutations, and genomic instability in gastric epithelial cells. Chronic inflammation also increases cell turnover, providing more opportunities for mutations to accumulate.
A) is incorrect H. pylori does not secrete exotoxins that directly damage DNA. Instead, its virulence factors disrupt cellular processes and promote inflammation.
B) is partially incorrect because although H. pylori infection can alter apoptosis, its primary carcinogenic mechanism is through chronic inflammation and DNA damage.
C) is incorrect as although some viruses (e.g., HPV) integrate their DNA into the host genome, this is not a mechanism used by H. pylori.
E) is incorrect.
Question 24:
Answer: C) It is an oxidative lesion that causes G:C to T:A transversions if unrepaired.
Explanation: 8-oxoG forms when guanine undergoes oxidation due to reactive oxygen species (ROS). If unrepaired, 8-oxoG mispairs with adenine during replication, leading to a G:C to T:A transversion, a mutagenic event. This lesion is repaired by base excision repair (BER).
A) is incorrect because depurination involves the loss of purine bases (adenine or guanine) from the DNA backbone. This does not involve 8-oxoG which is repaired by the Base Excision Repair (BER) pathway, not nucleotide excision repair (NER).
B) is incorrect as interstrand crosslinks (ICLs) involve covalent bonds between the two strands of DNA and are repaired by the Fanconi anaemia pathway and homologous recombination. 8-oxoG does not form interstrand crosslinks.
D) is incorrect because bulky adducts, such as those caused by UV-induced pyrimidine dimers or chemical exposure, are repaired by NER. 8-oxoG is a small oxidative lesion, not a bulky adduct.
E) is incorrect because intercalation involves molecules inserting themselves between base pairs, causing structural distortion. 8-oxoG does not intercalate; it is a direct modification of guanine.
Question 25:
Answer: C) Impaired transcription-coupled nucleotide excision repair.
Explanation: Cockayne syndrome is a rare disorder caused by defects in transcription-coupled NER, leading to photosensitivity, neurodegeneration, and premature aging. Cockayne syndrome results from mutations in CSA or CSB, proteins essential for transcription-coupled NER. This pathway specifically removes bulky lesions from actively transcribed genes to ensure proper transcription.
A) is incorrect because failure to repair bulky DNA adducts in non-transcribed regions of the genome is characteristic of xeroderma pigmentosum (XP), specifically in the global genome NER pathway. Cockayne syndrome primarily affects transcription-coupled NER.
B) is incorrect because single-strand break repair is handled by the BER pathway, involving PARP1 and XRCC1. This is unrelated to the defects in Cockayne syndrome.
D) is incorrect because mismatch repair (MMR) defects are seen in conditions like Lynch syndrome. Cockayne syndrome does not involve MMR defects.
E) is incorrect because homologous recombination defects are characteristic of BRCA1/2 mutations or Fanconi anaemia, not Cockayne syndrome.
Question 26:
Answer: B) Covalent DNA adduct formation repaired by nucleotide excision repair.
Explanation: PAHs in tobacco smoke are potent carcinogens due to their ability to form bulky DNA adducts, which, if unrepaired, lead to mutations and cancer development. PAHs are metabolised into reactive intermediates that form covalent adducts with DNA bases, particularly guanine. These bulky adducts distort the DNA helix and are repaired by the global genome NER pathway.
A) is incorrect because PAHs do not cause interstrand crosslinks. These kinds of lesions are typically caused by agents like cisplatin.
C) is incorrect because 8-oxoG is formed by oxidative stress, not by PAHs.
D) is incorrect because PAHs do not directly cause DNA methylation. This mechanism is more relevant to epigenetic changes in cancer.
E) is not the best answer as while PAHs can induce oxidative stress, their primary carcinogenic mechanism involves DNA adduct formation, not direct double-strand breaks.
Question 27:
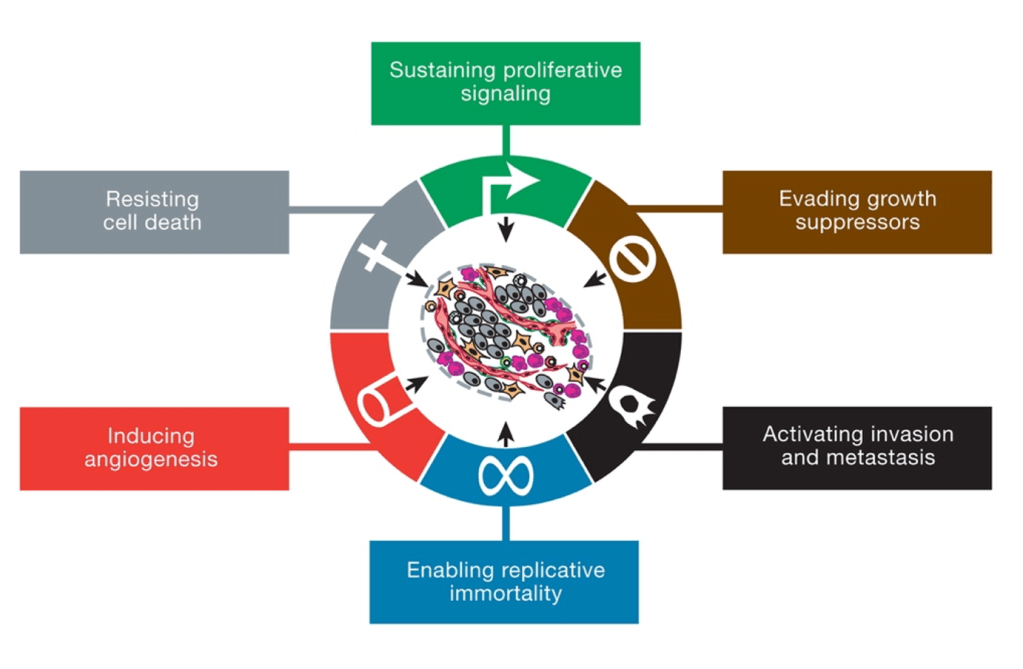
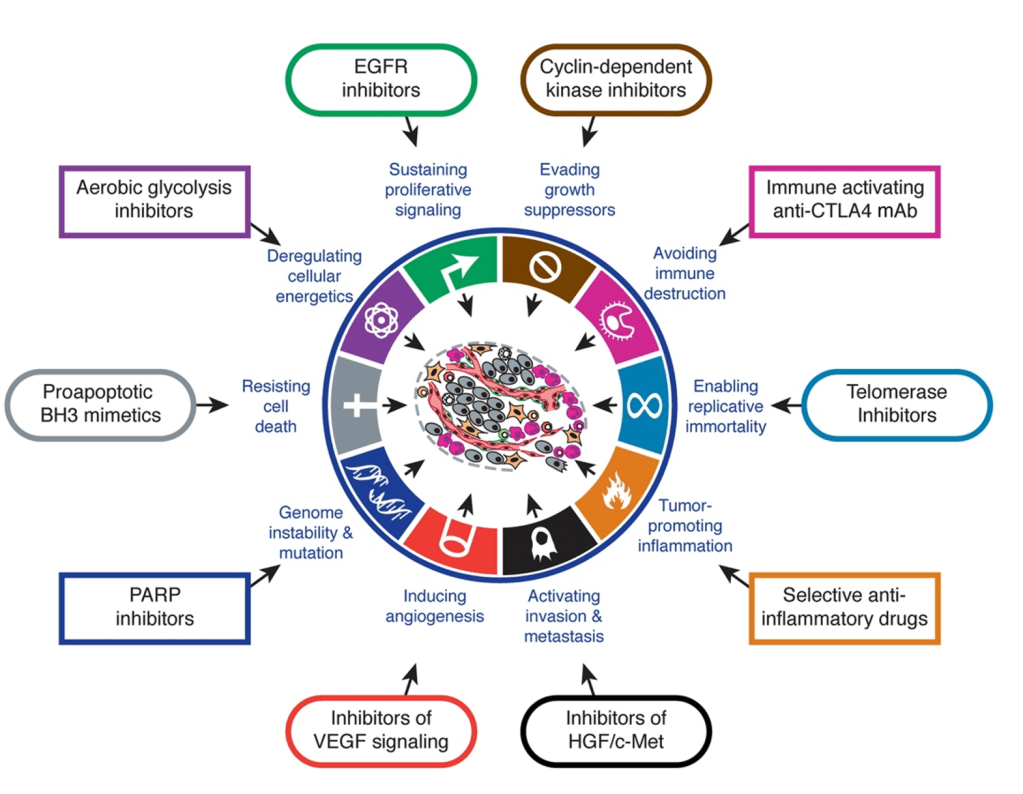
Answer: E) Avoiding immune destruction
Explanation: The six original hallmarks of cancer, as described by Hanahan and Weinberg in their seminal 2000 paper, include:
- Sustaining proliferative signalling
- Evading growth suppressors.
- Activating invasion and metastasis.
- Enabling replicative immortality.
- Inducing angiogenesis.
- Resisting cell death.
“Avoiding immune destruction” was introduced later in their updated 2011 paper as an emerging hallmark, not one of the six original hallmarks.
Option A is incorrect because “Sustaining proliferative signalling” is one of the original six hallmarks, describing how cancer cells continuously receive signals to proliferate or no longer requiring external signals such as due to KRAS mutations.
Option B is incorrect because “Evading growth suppressors” is an original hallmark, describing the ability of cancer cells to bypass mechanisms that normally inhibit cell growth such as TP53 & Rb.
Option C is incorrect because “Activating invasion and metastasis” is one of the original hallmarks, referring to cancer’s ability to spread to other tissues and organs.
Option D is incorrect because “Inducing angiogenesis” is an original hallmark, referring to the process by which cancer promotes the growth of blood vessels to sustain itself e.g. by secreting Vascular Endothelial Growth Factor VEG-F.
Question 28:
Answer: C) TP53
Explanation: High-grade serous ovarian carcinoma (HGSC) is the most common and aggressive subtype of epithelial ovarian cancer. This cancer is strongly associated with mutations in TP53, a tumour suppressor gene that regulates the cell cycle and prevents abnormal cell proliferation. TP53 mutations are the most frequently observed mutations across all human cancers, including ovarian cancer. In fact, more than 90% of high-grade serous ovarian carcinomas have TP53 mutations.
Option A is incorrect because BRCA1 mutations are associated with ovarian cancer, particularly in hereditary breast and ovarian cancer syndrome. However, TP53 mutations are more commonly found in high-grade serous ovarian carcinoma AND is the most commonly mutated gene in all human cancers.
Option B is incorrect because KRAS mutations are more frequently associated with pancreatic, colon, and lung cancers, and are not the most common mutations in ovarian cancers, especially high-grade serous ovarian carcinoma.
Option D is incorrect because PTEN mutations are more commonly linked to cancers of the endometrium, prostate, and breast, rather than high-grade serous ovarian carcinoma.
Option E is incorrect because EGFR mutations are typically found in cancers of the lung, head and neck, and other epithelial cancers, but they are not the most common mutations in ovarian cancer, particularly in the high-grade serous subtype.
Question 29:
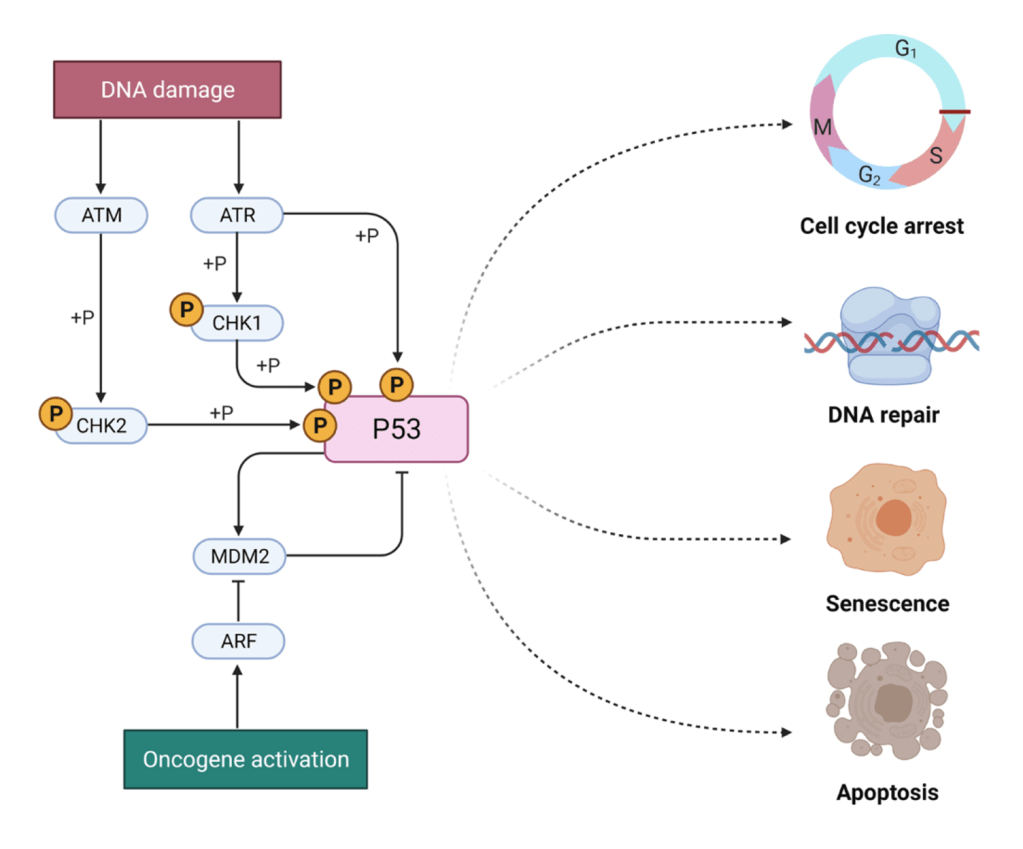
Answer: D) Unregulated phosphorylation of Rb, promoting uncontrolled progression from G1 to S phase.
Explanation: The TP53 gene encodes the p53 protein, a key tumour suppressor that regulates the cell cycle and apoptosis. Under normal conditions, p53 activates the expression of genes such as p21, which inhibits cyclin-dependent kinases (CDKs) namely CDK-2. This inhibition leads to the dephosphorylation of the retinoblastoma protein (Rb), which in turn halts the cell cycle at the G1 phase. However, when TP53 is mutated, this mechanism is disrupted. Without functional p53, p21 expression is impaired, allowing CDKs to phosphorylate Rb unchecked. This results in the release of E2F transcription factors, which drive the cell cycle from G1 to S phase, leading to uncontrolled progression through the cell cycle and potentially contributing to tumorigenesis.
Option A is incorrect because TP53 mutations impair the expression of p21. This prevents the normal inhibition of CDKs and leads to uncontrolled cell cycle progression rather than enhanced cell cycle arrest at G1.
Option B is incorrect because TP53 mutations impair the activation of apoptotic pathways. Normally, p53 induces the expression of pro-apoptotic genes (such as Bax) and caspases in response to cellular stress, but mutations in TP53 prevent this, leading to defective apoptosis rather than enhanced apoptotic resistance due to p53 loss of function.
Option C is incorrect because TP53 mutations do not lead to the upregulation of FAS receptors. FAS receptor-mediated apoptosis is part of the extrinsic apoptosis pathway, and p53 primarily regulates the intrinsic apoptosis pathway. Mutations in TP53 mainly impair the intrinsic pathway, not the extrinsic pathway.
Option E is incorrect because TP53 mutations prevent the normal function of p21. While p21 normally inhibits CDKs and arrests the cell cycle at G1, in the presence of a TP53 mutation, p21 levels are reduced, leading to continuous phosphorylation of Rb and unrestricted progression through the cell cycle.
Question 30:
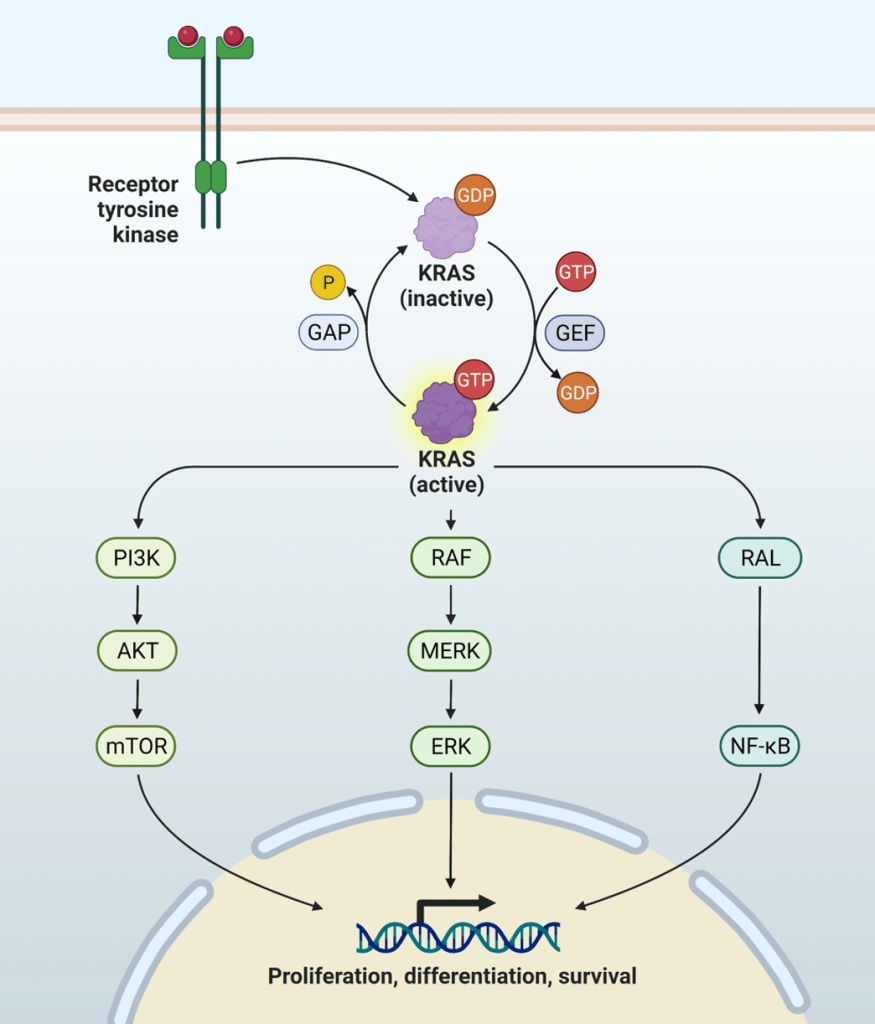
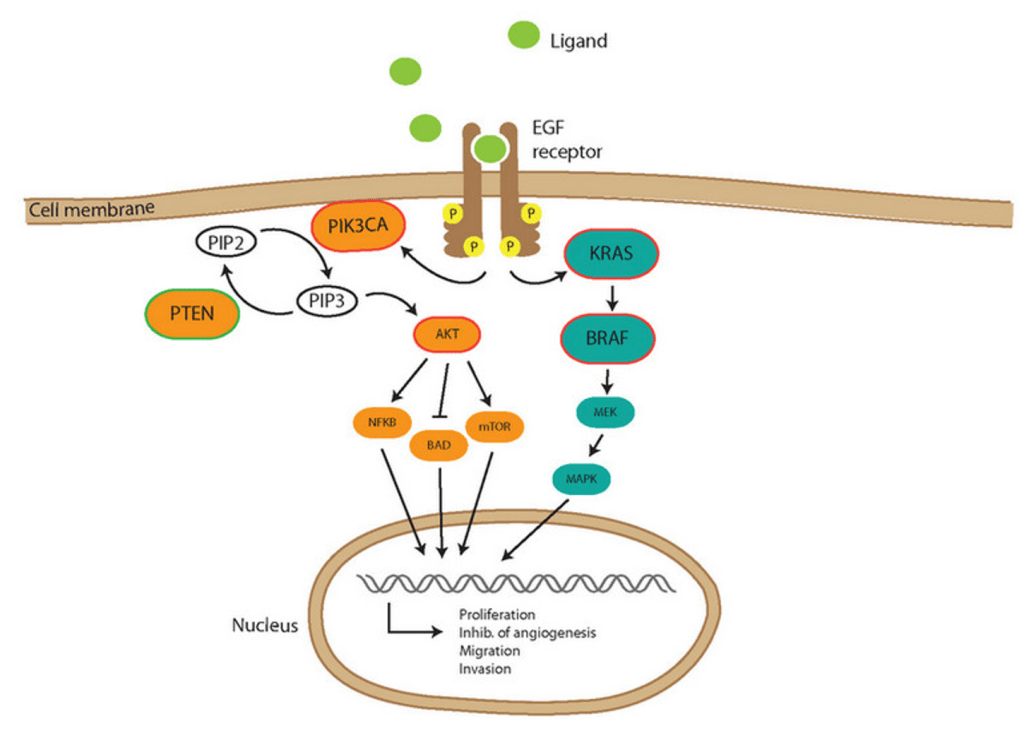
Answer: C) The KRAS mutation leads to the activation of downstream signalling proteins, such as RAF and MEK, which continuously promote cell cycle progression without requiring external growth factor signalling.
Explanation: KRAS is a key gene in cell signalling pathways that regulate growth and proliferation. When mutated, KRAS becomes constitutively active, meaning it is always “on” and continuously signals downstream pathways, such as RAF, MEK, and ERK. This leads to constant activation of signalling pathways that drive cell cycle progression and proliferation, independent of external growth signals such as growth factors. This allows cancer cells to sustain proliferative signalling even in the absence of normal growth factor stimulation. The KRAS mutation affects GAP, an enzyme that inactivates KRAS by hydrolysing GTP to GDP. When GAP is ineffective due to the KRAS mutation, KRAS remains in its active, GTP-bound state.
Option A is incorrect because the KRAS mutation does not directly cause the overproduction of growth factors like PDGF. Instead, it results in the continuous activation of signalling pathways that promote cell proliferation.
Option B is incorrect because the KRAS mutation does not upregulate growth factor receptors such as EGFR. Instead, it causes a change in the downstream signalling proteins that activate growth pathways irrespective of external signals.
Option D is incorrect because the KRAS mutation does not lead to the production of CSF-1 (colony-stimulating factor 1). While CSF-1 can influence the tumour microenvironment, it is not the direct mechanism by which KRAS sustains proliferative signalling.
Option E is incorrect because the KRAS mutation does not directly induce the degradation of tumour suppressor genes. The primary effect of the KRAS mutation is the constitutive activation of downstream signalling pathways, leading to uncontrolled cell proliferation.
Question 31:
Answer: D) Cancer cells downregulate tumour suppressor genes like p21 to enhance growth factor sensitivity.
Explanation: Sustaining proliferative signalling is a hallmark of cancer, and cancer cells achieve this through various mechanisms that ensure continuous activation of growth-promoting pathways. These include producing their own growth factors, increasing growth factor receptor expression, activating downstream signalling proteins, and influencing the tumour microenvironment to support their growth. However, the downregulation of tumour suppressor genes like p21 is not directly a mechanism for sustaining proliferative signalling; instead, it is related to evading growth suppressors.
Option A is incorrect because cancer cells do produce their own growth factors in an autocrine manner. For example, glioblastomas produce PDGF (platelet-derived growth factor), which stimulates their own growth.
Option B is incorrect because cancer cells increase the number of growth factor receptors on their surface to become hypersensitive to external growth signals. An example is the upregulation of EGFR (epidermal growth factor receptor).
Option C is incorrect because mutations in KRAS lead to constitutive activation of downstream signalling pathways like RAF-MEK-ERK, which promote continuous cell cycle progression and proliferation, even in the absence of external growth factors.
Option E is incorrect because cancer cells can influence their microenvironment to sustain growth signals. For instance, mammary cancer cells secrete CSF-1 (colony-stimulating factor 1) to recruit macrophages, which in turn secrete growth factors to support cancer cell proliferation.
Question 32:
Answer: E) Decreased expression of anti-apoptotic factors such as Bcl-2 & Bcl-XL is a key mechanism cancer cells use to evade apoptosis.
Explanation: Cancer cells evade apoptosis, a hallmark of cancer, by altering apoptotic pathways to resist programmed cell death. They often upregulate anti-apoptotic factors like Bcl-2 and Bcl-XL, rather than decrease their expression. Increased levels of these proteins help cancer cells inhibit mitochondrial outer membrane permeabilisation (MOMP), thereby blocking apoptosis and promoting survival. Option E is incorrect because decreased expression of anti-apoptotic factors like Bcl-2 and Bcl-XL would make cells more susceptible to apoptosis, not resistant.
Option A is incorrect because mutations in TP53 are a well-documented mechanism by which cancer cells impair the activation of pro-apoptotic proteins such as Bax, Bak, and Puma, thereby disabling the intrinsic apoptotic pathway.
Option B is incorrect because increased expression of Bcl-2 is a common mechanism used by cancer cells to inhibit mitochondrial outer membrane permeabilisation, thereby blocking the release of cytochrome c and preventing apoptosis.
Option C is incorrect because cancer cells can indeed produce non-signalling decoy FAS receptors, which bind Fas ligand and prevent activation of the extrinsic apoptotic pathway.
Option D is incorrect because production of insulin-like growth factors (IGF) enhances survival signalling pathways, such as the PI3K/AKT pathway, helping cancer cells resist cell death and evade apoptosis.
Question 33:
Answer: D) Cancer cells can only survive when located more than 500μm away from a blood vessel by activating alternative energy pathways.
Explanation: The statement in Option D is incorrect because cancer cells cannot survive when located more than 500μm away from a blood vessel unless they induce angiogenesis to establish a new blood supply. Cancer cells can only survive when they are 100μm away from a blood vessel; otherwise, they will die. Without access to sufficient oxygen and nutrients, cancer cells typically undergo necrosis or apoptosis. The angiogenic switch, which often involves upregulation of pro-angiogenic factors like VEGF-A, allows cancer cells to recruit blood vessels to sustain growth and survival. Cancer cells do not rely solely on alternative energy pathways to overcome oxygen and nutrient deprivation; instead, they stimulate vascularisation to ensure adequate supply.
Option A is incorrect because cancer cells evade the Hayflick limit (the normal restriction on the number of cell divisions) by upregulating telomerase, an enzyme that maintains telomere length and prevents chromosomal instability.
Option B is incorrect because telomerase is functionally absent in most healthy adult cells, limiting their replicative capacity. In contrast, telomerase is highly active in most cancer cells, allowing them to divide indefinitely and achieve replicative immortality.
Option C is incorrect because the angiogenic switch in cancer is frequently triggered by hypoxia (low oxygen levels). This leads to the activation of hypoxia-inducible factors (HIFs), which upregulate pro-angiogenic factors such as VEGF-A (vascular endothelial growth factor A).
Option E is incorrect because downregulation of anti-angiogenic factors like thrombospondin-1 (TSP-1) is a mechanism by which tumours promote vascularisation. Reduced expression of anti-angiogenic factors such as TSP-1 shifts the balance in favour of angiogenesis, enabling tumour growth.
Question 34:
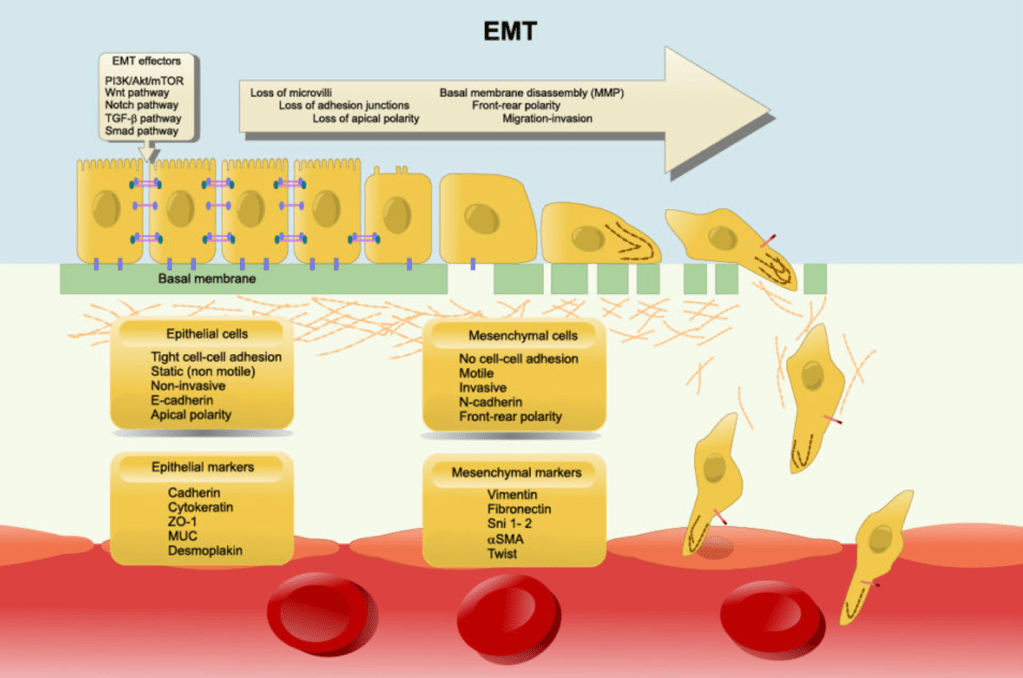
Answer: B) Induction of epithelial-mesenchymal transition (EMT), which reduces epithelial characteristics and enhances migratory and invasive capabilities.
Explanation: Metastasis is a complex process in which cancer cells spread from the primary tumour to distant sites. One of the central mechanisms that enable this process is epithelial-mesenchymal transition (EMT). During EMT, cancer cells lose their epithelial characteristics, such as tight cell-cell junctions and polarity, and acquire mesenchymal features that promote motility, invasiveness, and the ability to invade surrounding tissues. EMT reduces adhesion between cancer cells and enhances their ability to migrate through tissues and enter the bloodstream, which is crucial for metastasis. One of the main ways cancer cells promote metastasis is by decreasing E-Cadherin concentration & increasing N-Cadherin.
Option A is incorrect because overexpression of E-cadherin would actually promote increased adhesion between cells, which is not conducive to metastasis. In fact, loss of E-cadherin expression is a key feature of EMT and is necessary for cancer cells to become more mobile and invasive.
Option C is incorrect because increased synthesis of laminin in the extracellular matrix (ECM) would help stabilise the surrounding tissue and limit tissue invasion. For metastasis, cancer cells need to degrade and remodel the ECM to invade surrounding tissues and migrate to distant sites.
Option D is incorrect because suppression of N-cadherin would limit the ability of cancer cells to interact with stromal cells and reduce their invasiveness. N-cadherin is involved in cell-cell adhesion within tissues, and upregulation of N-cadherin can promote the invasive behaviour required for metastasis.
Option E is incorrect because enhanced stabilisation of cell polarity and cytoskeletal organisation would make it harder for cancer cells to become migratory. In fact, loss of polarity and changes in the cytoskeleton are part of the process of EMT, which allows cells to detach and migrate more easily.
Question 35:
Answer: D) The K-RasG12V mutation allows K-Ras to remain permanently activated, regardless of EGFR signalling, bypassing the effects of Cetuximab.
Explanation: The K-RasG12V mutation is a well-known activating mutation that results in the constitutive activation of the K-Ras protein. It is the MOST COMMON mutation affecting K-RAS. In its active form, K-Ras promotes downstream signalling through the MAPK and PI3K pathways, which are critical for cell proliferation and survival. The key feature of the K-RasG12V mutation is that it makes K-Ras continuously active, regardless of EGFR signalling. Therefore, even if Cetuximab inhibits EGFR, which is meant to block the upstream signal that activates K-Ras, the downstream signalling remains active due to the mutation. This is why the treatment shows no significant effect in this patient.
Option A is incorrect because while the K-RasG12V mutation causes K-Ras to be permanently active, it does not directly affect the EGFR itself. The mutation bypasses the need for EGFR signalling, but not by making EGFR permanently active.
Option B is incorrect because the K-RasG12V mutation does not cause EGFR receptor downregulation. Cetuximab targets the extracellular domain of the EGFR, and the mutation in K-Ras affects the downstream signalling, not the availability of EGFR itself.
Option C is incorrect because the K-RasG12V mutation results in the constitutive activation of K-Ras, not a deficiency in GTP binding. The mutation causes K-Ras to stay active even without GTP binding being properly regulated. The K-RAS mutations affects GAP, an enzyme that inactivates KRAS by hydrolysing GTP to GDP. When GAP is ineffective due to the KRAS mutation, KRAS remains in its active, GTP-bound state.
Option E is incorrect because the K-RasG12V mutation does not affect the binding affinity of Cetuximab. Cetuximab works by blocking EGFR signalling, but the mutation in K-Ras bypasses EGFR signalling entirely, rendering the inhibition ineffective.
Question 36:
Answer: A) Amplification of c-myc on chromosome 4 results in excessive production of the c-myc protein, driving uncontrolled cell proliferation.
Explanation: The c-myc gene is an oncogene that plays a central role in the regulation of cell growth, proliferation, and apoptosis. Amplification or translocation of the c-myc gene leads to its overexpression, resulting in an excess of c-myc protein. This overexpression of c-myc promotes uncontrolled cell proliferation by driving the cell cycle forward and by inhibiting apoptosis. The c-myc protein regulates the expression of genes involved in cell division and metabolism, and its dysregulation is a key event in the development of many cancers, including lymphoma, neuroblastoma, and other solid tumours.
Option B is incorrect because translocation of c-myc from chromosome 8 to chromosome 14 places it next to the immunoglobulin heavy chain (IgH) gene, leading to the overexpression of c-myc. This results in increased c-myc protein levels, which drive tumorigenesis, not inhibit it.
Option C is incorrect because c-myc amplification is not restricted to adult cancers. It plays a significant role in the development and prognosis of childhood cancers such as neuroblastoma, where c-myc amplification is associated with poor prognosis and more aggressive disease.
Option D is incorrect because HER2/Neu gene amplification is associated with breast cancer, but it does not directly result in the production of c-myc proteins. Instead, HER2 amplification leads to the activation of downstream signalling pathways such as the MAPK and PI3K pathways, which promote cell survival and proliferation. While c-myc may be upregulated as part of this process, it is not the direct cause of breast cancer metastasis.
Option E is incorrect because amplification of c-myc does not cause a reduction in c-myc protein levels. Instead, it causes overproduction of the c-myc protein, leading to enhanced cell proliferation and survival, which is a key mechanism of tumorigenesis.
Question 37:
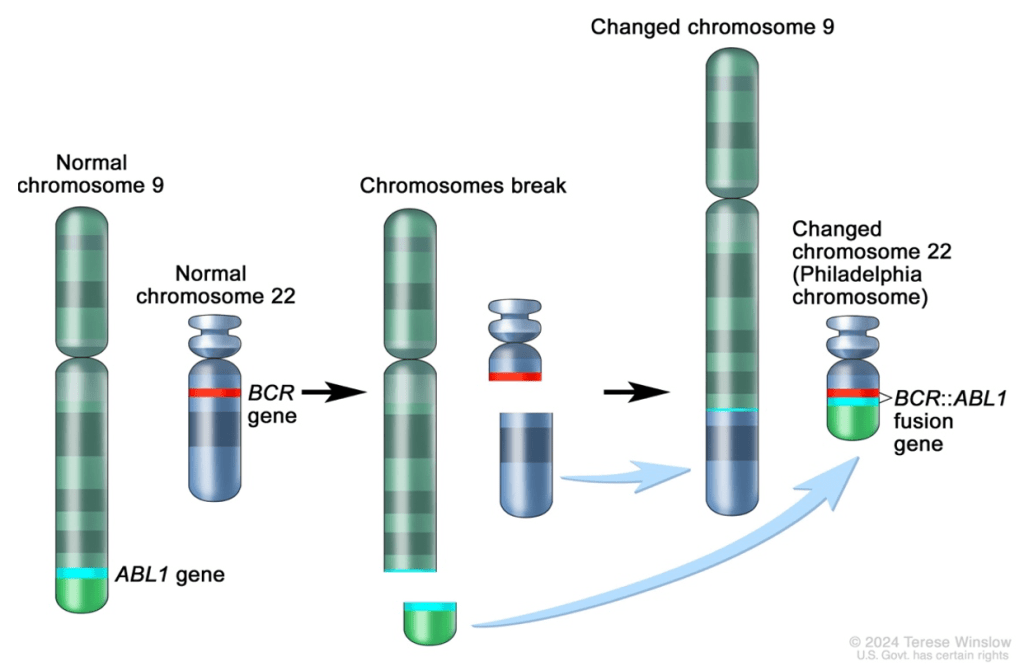
Answer: A) The translocation of ABL-1 to chromosome 22 next to BCR results in the formation of a hybrid BCR-ABL protein that encodes for a tyrosine kinase with enhanced activity, promoting uncontrolled cell division.
Explanation: The Philadelphia chromosome is a result of a translocation between chromosomes 9 and 22, specifically the ABL-1 gene on chromosome 9 and the BCR gene on chromosome 22. This translocation forms a hybrid BCR-ABL gene, which codes for a BCR-ABL fusion protein with enhanced tyrosine kinase activity. Normally, ABL-1 is a proto-oncogene that encodes a non-receptor tyrosine kinase involved in regulating cell growth and division. When BCR-ABL is formed, the fusion protein continuously activates downstream signalling pathways, including the Ras/Raf/MAPK and PI3K/Akt pathways, promoting uncontrolled cell proliferation and inhibiting apoptosis. This mechanism plays a central role in the pathogenesis of Chronic Myeloid Leukaemia (CML) and some cases of Acute Lymphoblastic Leukaemia (ALL).
Option B is incorrect because the Philadelphia chromosome does not downregulate BCR and ABL-1. Instead, the translocation results in the creation of a hybrid protein with increased tyrosine kinase activity, which drives cancer cell proliferation.
Option C is incorrect because the BCR-ABL translocation does not prevent the formation of tyrosine kinases; rather, it results in the production of a hyperactive tyrosine kinase. This altered protein enhances signalling that promotes cancer cell survival and growth.
Option D is incorrect because imatinib (a tyrosine kinase inhibitor) targets the BCR-ABL hybrid protein by inhibiting its activity, not enhancing it. By inhibiting BCR-ABL activity, imatinib blocks the uncontrolled cell proliferation characteristic of CML.
Option E is incorrect because the Philadelphia chromosome is not exclusive to ALL. It is also a key factor in Chronic Myeloid Leukaemia (CML), where it is found in nearly 95% of patients, in addition to being present in some cases of ALL.
Question 38:
Answer: B) If the viral genome inserts next to a gene such as c-myc, it can result in the amplification of that gene, leading to overproduction of the c-myc protein and promoting uncontrolled cell proliferation.
Explanation: Viral genome insertion can contribute to cancer development through a process called “cis-activation.” When a virus integrates its genome into the host DNA, the viral genome may insert near a proto-oncogene (such as c-myc). This insertion can cause the proto-oncogene to be overexpressed, often by promoting gene amplification or by altering the regulatory elements controlling the gene. In the case of c-myc, this results in excessive production of the c-myc protein, which drives uncontrolled cell proliferation and contributes to tumorigenesis.
Option A is incorrect because viral genome insertion does not typically inhibit tumour suppressor genes in a random manner. Instead, it tends to activate proto-oncogenes or alter the expression of key growth-regulating genes, contributing to cancer.
Option C is incorrect because viral insertion does not directly induce mutations in tumour suppressor genes to transform the host genome into a cancerous form. Rather, it is often the activation or overexpression of oncogenes (e.g., c-myc) that contributes to cancer, not mutations in tumour suppressor genes.
Option D is incorrect because while viral insertion can lead to the activation of proto-oncogenes, this is a specific mechanism of action. The viral genome itself does not directly cause mutations in proto-oncogenes; rather, it can enhance their expression or cause gene amplification, leading to uncontrolled growth.
Option E is incorrect because the viral genome does not replicate independently without affecting the host’s cellular processes. In fact, the insertion of the viral genome into the host DNA typically alters the host’s cellular machinery, promoting oncogene expression and disrupting normal cell cycle regulation.
Question 39:
Answer: B) c-myc amplification results in the continuous expression of MYC, which binds to MAX and activates numerous proliferative signalling pathways, since MAD can no longer bind & inhibit c-myc.
Explanation: The c-myc gene encodes a transcription factor that plays a key role in regulating cell growth, proliferation, and differentiation. When the c-myc gene is amplified, it leads to overproduction of the MYC protein. MYC normally forms a complex with the MAX protein, and this MYC-MAX complex activates genes involved in cell cycle progression and proliferation. The amplification of c-myc causes continuous expression of MYC, leading to persistent activation of these proliferative pathways. Additionally, the overexpression of MYC can prevent the binding of MAD (a MYC antagonist) to MAX, further enhancing proliferative signals, which contributes to uncontrolled cell division and tumorigenesis.
Option A is incorrect because amplification of c-myc does not cause the overproduction of MAD. MAD is typically involved in inhibiting MYC activity by binding to MAX, but in the case of c-myc amplification, MAD’s inhibitory effect is diminished, not overproduced.
Option C is incorrect because amplification of c-myc does not lead to the production of a truncated form of MYC. Instead, it leads to the overproduction of the full-length MYC protein, which disrupts normal cellular regulation and drives tumorigenesis.
Option D is incorrect because amplification of c-myc does not cause the loss of MAX expression. Rather, it leads to the overproduction of MYC, which continues to bind to MAX, forming the MYC-MAX complex that activates proliferative signals.
Option E is incorrect because amplification of c-myc does not activate tumour suppressor pathways; instead, it activates proliferative, oncogenic pathways, contributing to cancer development. The MYC-MAX complex is involved in promoting cell growth, not suppressing it.
Question 40:
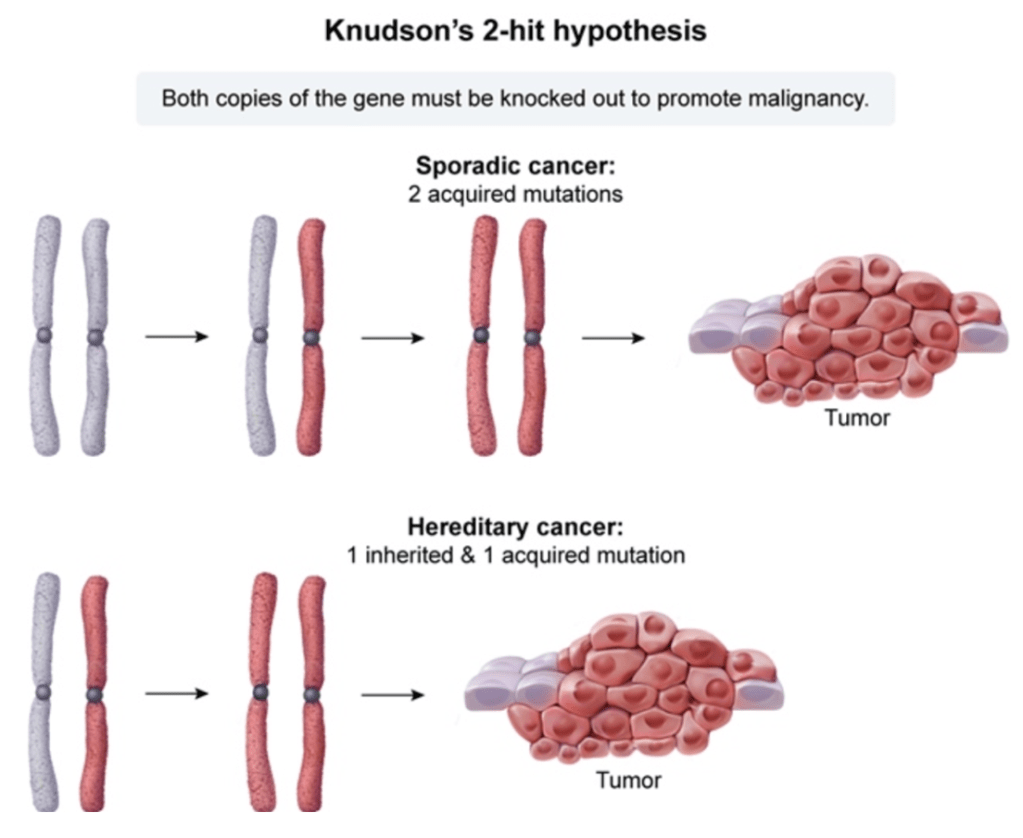
Answer: B) The child inherited a mutation in one RB1 allele, and a second, sporadic mutation occurred in the other allele in the affected eye, leading to tumour development.
Explanation: According to Knudson’s Two-Hit Hypothesis, the development of cancer in individuals with a genetic predisposition requires two “hits” or mutations to both alleles of a tumour suppressor gene. In the case of retinoblastoma, the RB1 gene is a tumour suppressor. Children with an inherited mutation in one allele of the RB1 gene are at a higher risk of developing retinoblastoma because the second, normal allele is still required to be inactivated to result in tumour formation. Typically, the second “hit” occurs later in life, either through a somatic mutation or loss of the remaining normal allele, which leads to the development of retinoblastoma.
Option A is incorrect because a single mutation in the RB1 gene is not sufficient to cause retinoblastoma. According to Knudson’s model, both alleles must be inactivated (i.e., both “hits” must occur) for the tumour to develop.
Option C is incorrect because this describes bilateral retinoblastoma, where both eyes are affected. In this case, the child has only one mutated RB1 allele and is at risk for developing retinoblastoma in one eye, not necessarily both. Bilateral retinoblastoma occurs when the child inherits two mutated alleles, one from each parent, or has mutations in both alleles from the start.
Option D is incorrect because a somatic mutation typically leads to the inactivation of the remaining normal allele in the affected eye, not in the unaffected eye. The development of retinoblastoma is typically unilateral in children who inherit one mutated allele and then acquire a second mutation in the affected eye.
Option E is incorrect because this describes a situation where both alleles are mutated during mitosis in the unaffected eye. However, the Two-Hit Hypothesis implies that mutations occur in the affected eye and the second “hit” happens after birth, not during mitosis in the unaffected eye. This would not typically result in bilateral retinoblastoma.
Question 41:
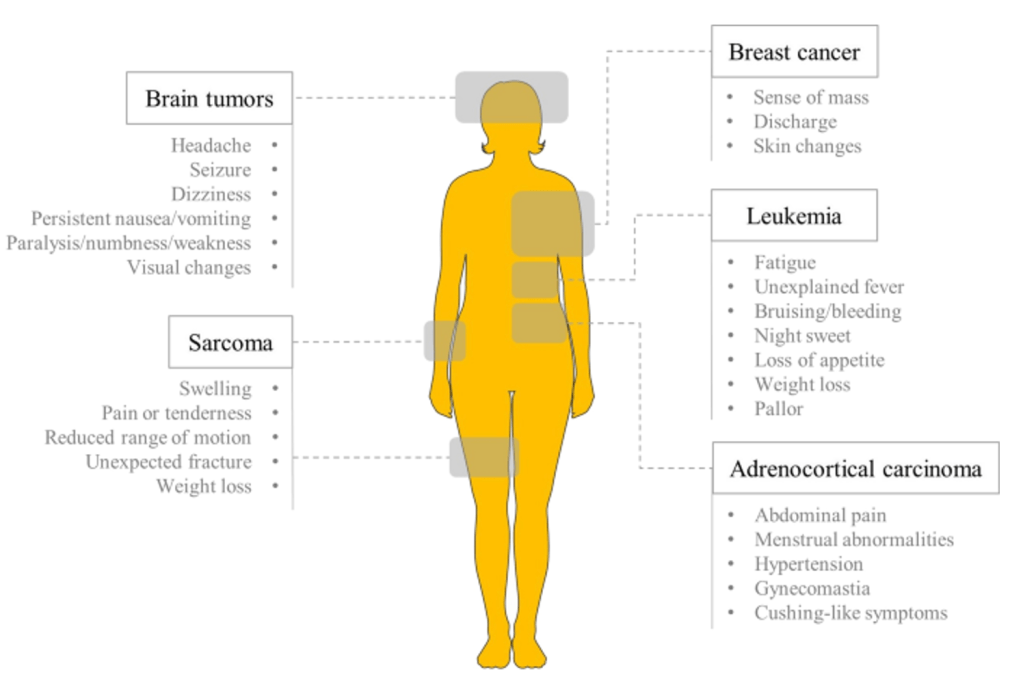
Answer: B) The likelihood of developing invasive cancer by 30 years old is 50%, and 90% by 70 years old.
Explanation: Li-Fraumeni Syndrome (LFS) is a hereditary condition caused by germline mutations in the p53 tumour suppressor gene, which significantly increases the risk of developing various types of cancer, including sarcomas, breast cancer, brain tumours, and adrenocortical carcinomas. Individuals with LFS have a much higher cancer risk compared to the general population. The likelihood of developing invasive cancer by age 30 is approximately 50%, and by age 70, the cumulative cancer risk is around 90%. This makes LFS one of the most high-risk genetic cancer syndromes.
Option A is incorrect because the cancer risk in individuals with Li-Fraumeni Syndrome is significantly higher than that of the general population, not 30% by age 30. The risk is about 50% by age 30.
Option C is incorrect because mutations in the p53 gene can occur both as inherited (germline) mutations and as somatic mutations. In Li-Fraumeni Syndrome, the mutation is inherited, but somatic mutations in p53 also play a significant role in tumorigenesis.
Option D is incorrect because Li-Fraumeni Syndrome is inherited in an autosomal dominant manner. Only one copy of the mutated gene is sufficient to predispose individuals to cancer. It does not require two mutations in the p53 gene for cancer to develop.
Option E is incorrect because individuals with Li-Fraumeni Syndrome are at risk for developing multiple types of cancers throughout their lifetime. It is not limited to a single cancer type.
Question 42:
Answer: C) APC mutations lead to persistent activation of beta-catenin, promoting uncontrolled cellular proliferation in the colon crypts.
Explanation: The APC (Adenomatous Polyposis Coli) gene is a key tumour suppressor gene involved in the regulation of the Wnt/β-catenin signalling pathway. In the normal colon, APC binds to β-catenin and promotes its degradation, preventing its accumulation in the cytoplasm. When APC is mutated, this degradation is inhibited, leading to the accumulation of β-catenin in the nucleus. In the nucleus, β-catenin activates the transcription of genes that promote cell proliferation, thus contributing to the early stages of colorectal cancer (CRC). This persistent activation of β-catenin is a crucial step in the development of colorectal adenomas, which can progress to carcinoma.
Option A is incorrect because the APC mutation does not cause excessive degradation of β-catenin. Rather, it prevents β-catenin degradation, leading to its accumulation and activation of proliferative genes.
Option B is incorrect because the APC mutation does not prevent Wnt ligand from binding to stem cells. Instead, the mutation leads to the dysregulation of the Wnt signalling pathway, resulting in persistent activation of β-catenin.
Option D is incorrect because APC mutations do not directly inactivate TGF-β signalling. The role of APC mutations is more directly related to the Wnt/β-catenin pathway rather than directly modulating TGF-β signalling. TGF-β signalling is another mutation that occurs later on that triggers the development of carcinoma.
Option E is incorrect because APC mutations do not directly inactivate p53 signalling. While p53 mutations are commonly involved in later stages of CRC progression, the APC mutation primarily affects β-catenin accumulation and subsequent cell proliferation rather than directly inactivating p53 signalling.
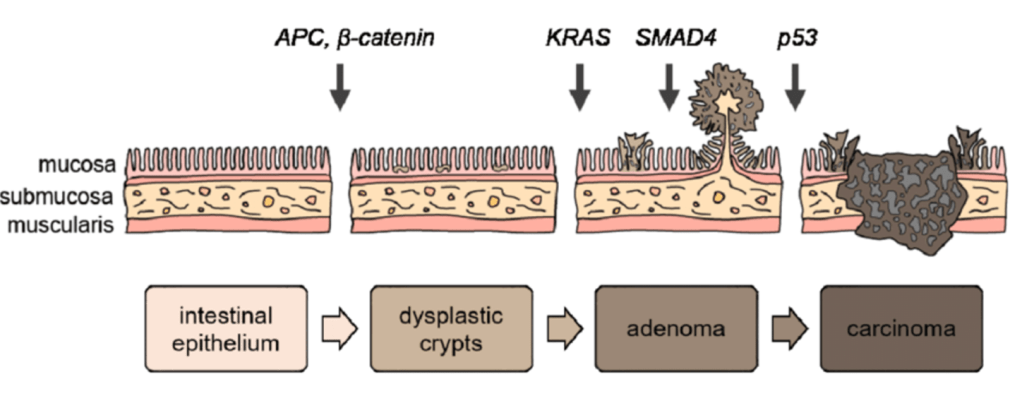
Question 43:
Answer: B) The mutation disrupts the recruitment of the Type I TGF-β receptor, impairing the activation of Smad2 and preventing the apoptosis-inducing effects of TGF-β signalling.
Explanation: TGF-β (Transforming Growth Factor-beta) is a critical regulator of cell growth, differentiation, and apoptosis in epithelial cells. The normal signalling pathway begins when TGF-β binds to its Type II receptor, which recruits and activates the Type I receptor. The activated Type I receptor then phosphorylates Smad2 (and Smad3), which forms a complex with Smad4. This complex translocates to the nucleus, where it regulates the transcription of genes that control cell-cycle arrest and apoptosis, particularly in response to cellular stress or DNA damage.
In this scenario, the mutation in the TGF-β receptor II gene disrupts the recruitment of the Type I TGF-β receptor. This prevents the phosphorylation of Smad2, blocking the activation of the Smad2/Smad4 complex. As a result, the downstream effects of TGF-β signalling are impaired, including the induction of cell-cycle arrest and apoptosis. This contributes to the inability of the cells to undergo normal growth control, promoting tumour progression.
Option A is incorrect because the mutation does not lead to persistent activation of the Smad2/Smad4 complex. Instead, it impairs the normal activation of this complex.
Option C is incorrect because the mutation does not result in enhanced secretion of TGF-β. It specifically disrupts the signalling process.
Option D is incorrect because the mutation does not cause the accumulation of the Type I receptor or promote the sustained activation of pro-apoptotic genes. It impairs receptor signalling.
Option E is incorrect because the mutation prevents the phosphorylation of Smad2, not as a result of a defect in the formation of the Smad2/Smad4 complex, but because it blocks the activation of the Type I receptor, which is necessary for Smad2 phosphorylation.
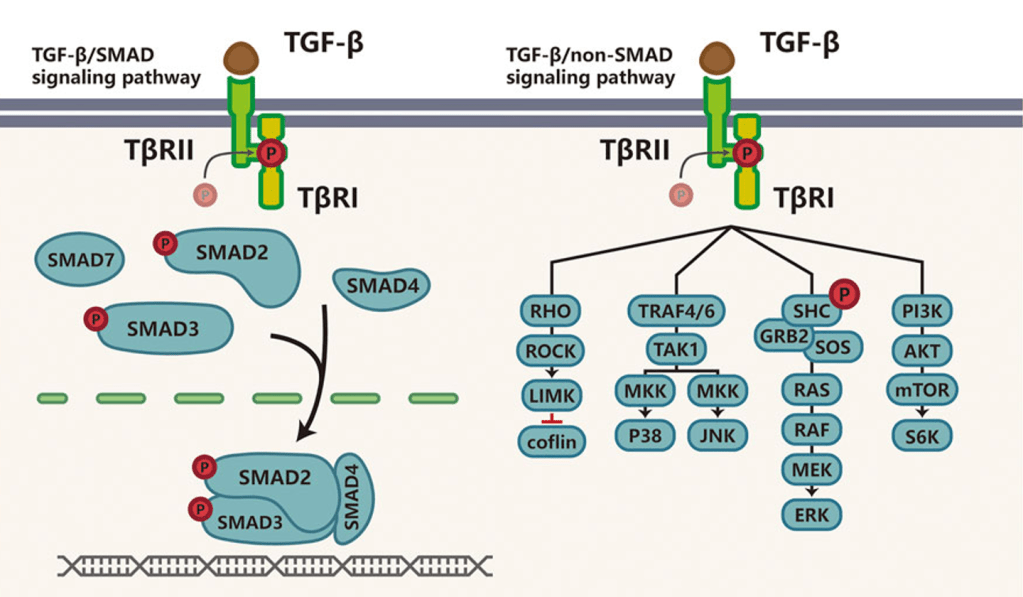
Question 44:
Answer: A) Familial Adenomatous Polyposis (FAP)
Explanation: Familial Adenomatous Polyposis (FAP) is a hereditary condition caused by mutations in the APC (Adenomatous Polyposis Coli) gene, which is a tumour suppressor gene. This mutation leads to the formation of numerous adenomatous polyps in the colon, often starting at a young age. These polyps have a high potential to progress to colorectal cancer if left untreated, typically by the fourth decade of life. The APC gene mutation disrupts the regulation of the Wnt signalling pathway, which is important for cell proliferation and differentiation in the colon. This results in uncontrolled cell growth and the formation of polyps that can later develop into cancer.
Option B is incorrect because Hereditary Non-Polyposis Colorectal Cancer (HNPCC), also known as Lynch syndrome, is caused by mutations in DNA mismatch repair genes (such as MLH1, MSH2, MSH6, and PMS2), not the APC gene. This condition leads to an increased risk of colorectal cancer but does not typically present with multiple adenomatous polyps.
Option C is incorrect because sporadic colorectal cancer is typically caused by a combination of environmental factors and random genetic mutations, not a hereditary mutation like the one seen in FAP. Sporadic colorectal cancer does not usually present with numerous polyps in the early stages.
Option D is incorrect because Lynch syndrome (Hereditary Non-Polyposis Colorectal Cancer) is caused by mutations in mismatch repair genes, as described above, and does not involve mutations in the APC gene.
Option E is incorrect because Gardner’s Syndrome is a variant of FAP that includes extra-intestinal manifestations such as osteomas, epidermoid cysts, and fibromas in addition to multiple colon polyps. However, the key diagnosis based solely on the APC mutation in this case is Familial Adenomatous Polyposis (FAP), which can also present with multiple adenomatous polyps.
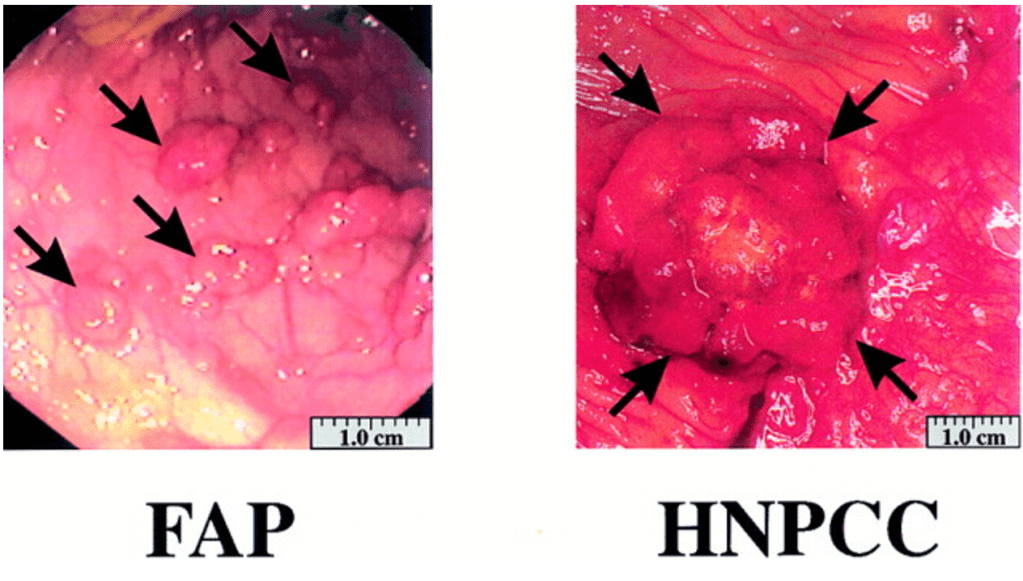
Question 45:
Answer: A) Methylation of CpG islands in the promoter region, preventing transcription factor binding
Explanation: Methylation of CpG islands in the promoter region of genes typically results in gene repression by altering the accessibility of DNA. The addition of a methyl group (by DNA methyltransferase) to the cytosine residue in CpG dinucleotides can inhibit the binding of transcription factors and other necessary regulatory proteins, thus preventing the initiation of transcription. This modification is a key mechanism in the silencing of genes, including in processes such as X-inactivation and the silencing of tumour suppressor genes in cancer.
Option B is incorrect because acetylation of histone proteins (by histone acetylase) is associated with gene activation, not repression. Acetylation neutralises the positive charge on histones, leading to a looser chromatin structure (euchromatin), which enhances DNA accessibility for transcription factors and promotes gene expression.
Option C is incorrect because phosphorylation of histone H3 at serine 10 is often linked to gene activation, not repression. This modification can lead to chromatin relaxation and is generally associated with the transcriptional activation of genes, particularly during the early phases of transcription.
Option D is incorrect because methylation of histone H3 at lysine 9 (H3K9me) is associated with the formation of heterochromatin, which represses gene expression. Heterochromatin is a tightly packed form of DNA that is not easily accessible for transcription, thus inhibiting transcription. However, H3K9me generally promotes gene repression rather than euchromatin formation.
Option E is incorrect because ubiquitination of histone H2A can either promote or inhibit transcription, depending on the specific context. In many cases, ubiquitination of histone H2A is linked to the repression of gene expression, particularly in the regulation of chromatin structure.
Question 46:
Answer: E) Dacogen & Vidaza
Explanation: Dacogen (decitabine) and Vidaza (azacitidine) are both nucleoside analogues that target the epigenome by inhibiting DNA methyltransferase activity. These drugs work by incorporating into DNA during replication, where they inhibit the DNA methyltransferase enzymes responsible for adding methyl groups to cytosine residues in CpG islands. This inhibition leads to hypomethylation of DNA and can reactivate silenced genes, including tumour suppressor genes, contributing to the restoration of normal gene expression in cancer cells. This mechanism is useful in treating cancers like myelodysplastic syndromes and acute myeloid leukaemia (AML), where epigenetic silencing plays a key role in the tumorigenesis.
Drugs which target epigenetic changes are good at selectively targeting cancerous cells. This is because healthy cells have epigenetic regulation; they are multi-layered and would require many epigenetic changes to occur before it would cause cancerous changes. However, cancerous cells are epigenetically more vulnerable – a slight tip in balance might push these cells into crisis. It is thought that haematological malignancies are more vulnerable to these therapies than solid tumours (mainly due to acquiring more mutations & having a more impressive tumour microenvironment).
Option A is incorrect because Vorinostat is a histone deacetylase inhibitor (HDACi) that targets histone modifications rather than DNA methylation to alter gene expression.
Option B (Depsipeptide) is incorrect because Depsipeptide is also a histone deacetylase inhibitor that modifies histone acetylation rather than DNA methylation.
Option C (Dacogen) is incorrect on its own, as while Dacogen does inhibit DNA methyltransferases, Vidaza (azacitidine) also has this same activity, making option E the best answer.
Option D (Vidaza) is incorrect on its own for the same reason as Dacogen. Both drugs work via the same mechanism of inhibiting DNA methyltransferase activity, and the correct answer includes both.
Question 47:
Answer: C) High Tumour-Associated Macrophage (TAM) infiltration
Explanation: Tumour-Associated Macrophages (TAMs) are a major component of the tumour immune microenvironment and are often associated with a poor prognosis in solid tumours due to their role in immune evasion and promoting tumour progression. TAMs can adopt a pro-tumour M2 phenotype that facilitates angiogenesis, tissue remodelling, and suppression of anti-tumour immunity, including inhibition of T-cell activity. TAMs can also secrete cytokines and growth factors that promote tumour cell proliferation, metastasis, and immune suppression, contributing to the overall aggressive nature of the tumour.
Option A is incorrect because high CD8+ T-cell infiltration is generally associated with a better prognosis, as CD8+ T-cells are cytotoxic and can target and kill tumour cells effectively.
Option B is incorrect because high CD4+ T-cell infiltration, particularly of Th1 cells, is often a sign of an effective immune response against tumours. However, the overall effect depends on the subset of CD4+ T-cells present.
Option D is incorrect because high Natural Killer (NK) cell activity is typically beneficial for tumour control, as NK cells are involved in the recognition and killing of tumour cells without prior sensitization.
Option E is incorrect because low T-Regulatory (T-Reg) cell presence is generally associated with a better prognosis. T-regs suppress immune responses and their presence in the tumour microenvironment can promote immune evasion; thus, low levels of T-regs would likely indicate a more effective anti-tumour immune response.
Question 48:
Answer: B) CAFs recruit stromal cells from the surrounding tissue to form the tumour stroma, which can increase interstitial pressures and limit drug perfusion.
Explanation: Cancer-Associated Fibroblasts (CAFs) play a crucial role in tumour progression by modifying the tumour microenvironment. They contribute to tumour growth and metastasis by secreting cytokines, growth factors, and extracellular matrix (ECM) components, which promote angiogenesis, immune evasion, and drug resistance.
Option A is incorrect because CAFs generally promote tumour progression rather than suppression, often by creating an immunosuppressive environment.
Option C is incorrect because CAFs do not enhance drug efficacy; rather, they can contribute to drug resistance by altering tumour metabolism and reducing drug penetration.
Option D is incorrect because CAFs are involved throughout all stages of tumour progression, including metastasis.
Option E is incorrect because the presence of CAFs is associated with poor prognosis, as they enhance tumour invasiveness and therapy resistance.
Question 49:
Answer: C) CAFs generally only tend to be involved in early stages of tumour development, before metastasis occurs contributing to tumour progression.
Explanation: Cancer-Associated Fibroblasts (CAFs) play a crucial role in tumour progression, not just in the early stages but also throughout tumour development, including during metastasis. CAFs contribute to tumour growth by modifying the extracellular matrix (ECM), promoting angiogenesis, and facilitating immune evasion. They support tumour cells and create a pro-tumour environment through the secretion of cytokines, growth factors, and ECM remodelling enzymes. Their role extends through all stages of tumour progression, including metastasis.
Option A is correct because CAFs can contribute to tumour progression by modifying the tumour stroma, increasing interstitial pressure, and creating physical barriers that hinder drug delivery, thereby reducing treatment efficacy.
Option B is correct because CAFs are known to express a variety of metabolic enzymes, including CYP3A4, which can metabolise and degrade chemotherapeutic agents, thereby reducing the efficacy of treatment.
Option D is correct because CAFs can influence immune cells within the tumour microenvironment and help tumours evade immune surveillance by inhibiting the activation of cytotoxic T cells, thereby promoting immune tolerance to the tumour.
Option E is correct because CAFs are indeed associated with poor prognosis. Higher levels of stromal invasion and fibrosis correlate with tumour aggressiveness, as CAFs promote tumour progression and reduce drug penetration, complicating treatment strategies.
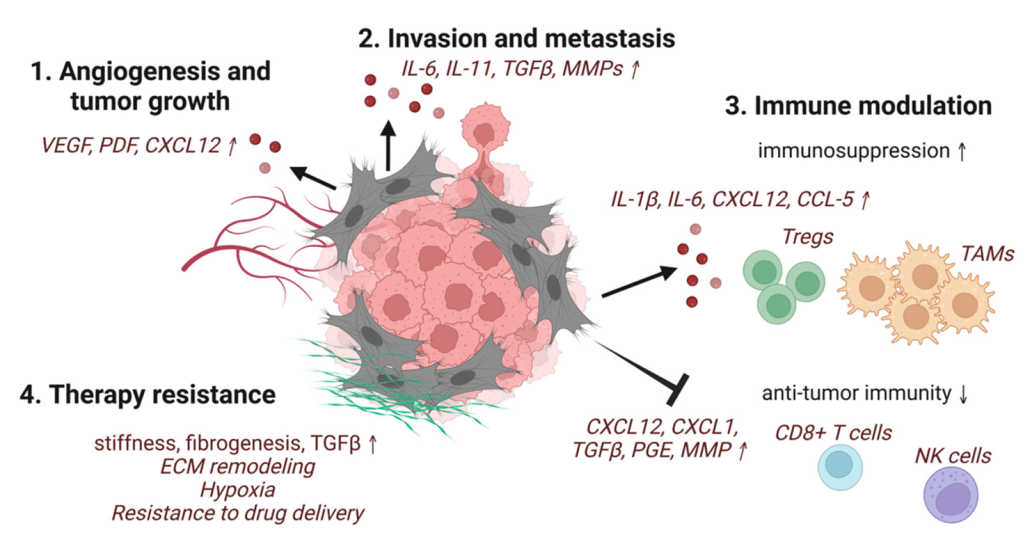
Question 50:
Answer: E) Tumours suppress T-cell-mediated immunity by upregulating the expression of co-stimulatory molecules, such as CD28, on CD8+ T-cells, promoting their activation.
Explanation: Tumours generally evade immune surveillance by employing mechanisms that suppress or inhibit T-cell activity. Option E is the least likely to contribute to immune evasion because upregulating co-stimulatory molecules like CD28 would promote T-cell activation (not suppress it), which is contrary to the immune evasion strategies employed by tumours. Tumours typically avoid immune detection by inhibiting T-cell activation, not enhancing it.
Option A is correct because tumours can recruit immunosuppressive cells like Tregs, MDSCs, and AAMs to suppress CD8+ T-cell activity and promote immune tolerance in the tumour microenvironment. The cells in the tumour microenvironment act in a bidirectional manner. Cancer cells secrete cytokines which influences the type of cells in the TME. Similarly, cells in the TME also secrete cytokines that trigger cancer cell progression & metastasis whilst simultaneously protecting the tumour from the immune response.
Option B is correct because many tumours lose the ability to present antigens effectively by mutating components of the MHC Class I processing pathway, such as the Tapasin complex, which reduces antigen presentation and helps the tumour escape immune surveillance by CD8+ T-cells.
Option C is correct because tumours can inhibit T-cell extravasation and infiltration by secreting factors that increase fibrosis and release endothelin, which blocks the entry of T-cells into the tumour site.
Option D is correct because tumour cells can engage immune checkpoint proteins like PD-1/PD-L1 to induce T-cell exhaustion and inhibit T-cell activation, thus promoting immune tolerance and evasion.
Question 51:
Answer: D) E. coli
Explanation: E. coli, specifically certain pathogenic strains like those producing colibactin, is particularly important in the development of mucous-associated colorectal cancer (CRC). E. coli can adhere to and invade epithelial cells, produce genotoxins such as colibactin, and stimulate pro-inflammatory and pro-angiogenic responses, which are all critical processes in promoting CRC. Colibactin, a genotoxin produced by certain E. coli strains, causes DNA damage in host cells, contributing to the development of cancer.
Option A is incorrect because Streptococcus gallolyticus is associated with CRC but is primarily linked to its ability to promote inflammation rather than direct genotoxicity.
Option B is incorrect because Bacteroides fragilis is known for producing a toxin that induces inflammation, but it is not as directly involved in the genotoxic processes as E. coli.
Option C is incorrect because Enterococcus faecalis is linked to CRC due to its ability to produce reactive oxygen species, but its role is not as prominent in the direct genetic damage associated with cancer development.
Option E is incorrect because Lactobacillus acidophilus is a beneficial bacterium and does not play a role in the development of CRC.
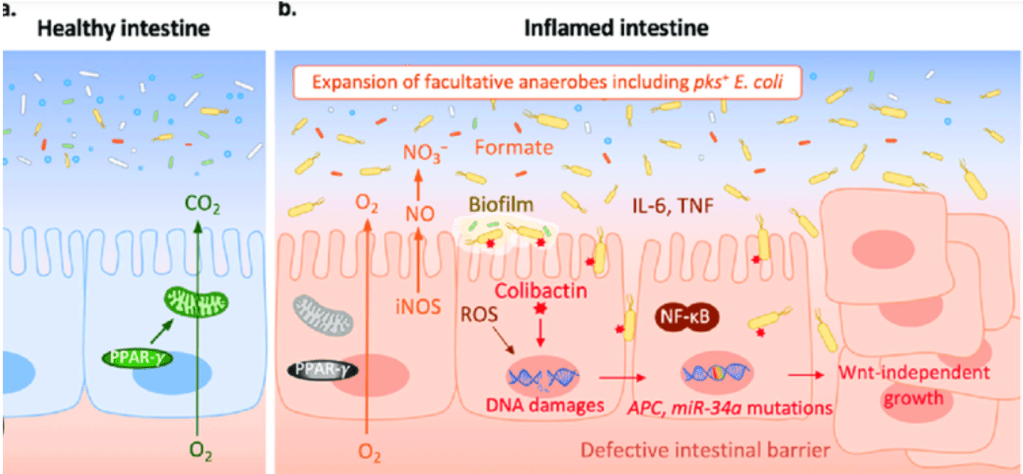

Question 52:
Answer: B) The reduction of Bacteroides and Firmicutes, along with an increase in Proteobacteria such as E. coli, promotes inflammation by stimulating Toll-like receptors (TLRs), leading to higher cellular proliferation and an increased risk of dysplasia and cancer.
Explanation: Dysbiosis, which is an imbalance in the gut microbiota, often involves a reduction in beneficial bacteria like Bacteroides and Firmicutes, and an increase in potentially harmful bacteria like Proteobacteria, including E. coli. This shift promotes inflammation by activating Toll-like receptors (TLRs) on epithelial cells, which in turn stimulates cellular proliferation. This heightened cellular turnover increases the risk of dysplasia, a precancerous condition, and subsequently colorectal cancer.
Option A is incorrect because the presence of more Firmicutes and fewer Proteobacteria would not typically increase immune activation or contribute to inflammation and cancer progression. In fact, higher levels of firmicutes & bifidobacteria(good bacteria) & lower levels of proteobacteria (harmful bacteria) would decrease inflammation & dysbiosis as this is the composition of a healthy colon.
Option C is incorrect because pro-inflammatory cytokines released from bacteria usually promote tumour growth by stimulating inflammation, rather than suppressing it.
Option D is incorrect because while genotoxins produced by bacteria like E. coli can be pro-carcinogenic, they are capable of directly affecting epithelial cell DNA, despite mucosal layers.
Option E is incorrect because while bacterial overgrowth may lead to polyps, a thickened mucosa does not prevent bacterial adhesion. In fact, increased bacterial presence & inflammation results in a thinner mucosa on top of polyps, not thicker which promotes adherence of bacteria & cancer progression.
Question 53:
Answer: C) CagA cytotoxin, which is injected into epithelial cells and influences signalling pathways linked to cancer progression.
Explanation: The CagA cytotoxin is considered the most important virulence factor of Helicobacter pylori (H. pylori) that contributes to its increased risk of gastric cancer. CagA is injected into gastric epithelial cells, where it disrupts normal cellular signalling pathways, leading to inflammation, abnormal cell growth, and alterations in the cell cycle. These changes increase the risk of gastric cancer by promoting carcinogenic processes. If you are infected with a CagA +ve strain, (which are also more common), the risk of gastric cancer increases x3.5.
Option A is incorrect because OipA adhesion facilitates binding to the gastric epithelium, but it is not directly linked to the development of gastric cancer. It primarily plays a role in the initial colonisation of the stomach, specifically the corpus.
Option B is incorrect because VacA is a cytotoxin that causes cellular damage, but its role in gastric cancer development is secondary to CagA. It is primarily involved in immune evasion and tissue damage rather than direct carcinogenesis. VacA is acytotoxin which is largely ubiquitous to all strains of H. Pylori.
Option D is incorrect because BabA adhesion is important for enhancing H. pylori colonisation, but it does not directly influence cancer risk.
Option E is incorrect because while SabA adhesion contributes to persistent colonisation, it does not have a direct impact on cancer progression.

Question 54:
Answer: B) HPV-related SCCHN is characterised by the inactivation of p53 by the viral E6 protein, which binds to p53 and tags it for degradation, allowing unchecked cell proliferation.
Explanation: The molecular pathogenesis of Human papillomavirus (HPV)-related squamous cell carcinoma of the head and neck (SCCHN) involves the viral E6 protein, which inactivates the tumour suppressor protein p53. By binding to p53 and tagging it for degradation, E6 prevents p53 from inducing cell cycle arrest or apoptosis, allowing for unchecked cell proliferation and the accumulation of genetic mutations that drive cancer development.
Option A is incorrect because while p53 is involved in HPV-related carcinogenesis, the primary mechanism is the inactivation of p53 by the E6 protein, not mutations in the p53 gene itself. Virally caused cancers do NOT cause mutations in p53 as most non-virally caused cancers do.
Option C is incorrect because the CDKN2A gene, which encodes the p16 protein, is associated with cell cycle regulation, but it is not the primary mechanism of carcinogenesis in HPV-related SCCHN. The inactivation of p53 by E6 is the key event.
Option D is incorrect because the overexpression of E2Fs and the binding of Rb by E7 also contribute to HPV-related cancer, however the mechanism is not entirely correct. E7 competitively binds to Rb preventing it from binding to E2Fs. This allows E2Fs to progress through the cell cycle & promote cell proliferation.
Option E is incorrect because while the presence of E6 and E7 is associated with carcinogenesis, the prognosis of HPV-positive SCCHN is generally better than HPV-negative SCCHN, not worse.
Question 55:
Answer: C) Cancer cells with a BRCA1 mutation cannot repair DNA via homologous recombination, so PARP inhibitors prevent the backup DNA repair mechanism, leading to synthetic lethality.
Explanation: Cancer cells with a BRCA1 mutation are unable to effectively repair DNA damage via homologous recombination (HR), a high-fidelity DNA repair mechanism. When treated with PARP inhibitors, the cells’ ability to repair DNA damage through an alternative pathway, such as base excision repair (BER) mediated by PARP, is blocked. This leads to the accumulation of DNA damage and ultimately results in cell death due to synthetic lethality. The loss of both DNA repair mechanisms—HR due to BRCA1 mutation and BER due to PARP inhibition—causes irreparable DNA damage that is lethal to the cancer cells.
Option A is incorrect because cancer cells with BRCA1 mutations typically rely on homologous recombination rather than non-homologous end joining (NHEJ) for accurate DNA repair. NHEJ is more error-prone.
Option B is incorrect because PARP inhibitors are not toxic to healthy cells due to inhibition of BRCA1; rather, they exploit the synthetic lethality between PARP inhibition and BRCA1 deficiency in cancer cells.
Option D is incorrect because PARP inhibitors do not block DNA ligase function. PARP inhibitors block the PARP enzyme itself, which is involved in repairing single-strand DNA breaks via base excision repair, not DNA ligases used in NHEJ.
Option E is incorrect because PARP inhibitors do not activate homologous recombination; they block an alternative repair pathway (BER), which, in the context of BRCA1 deficiency, leads to synthetic lethality.
Question 56:
Answer: B) The tumour has impaired antigen presentation due to the loss of MHC Class I expression, preventing the recognition of tumour-specific antigens by CD8+ T-cells.
Explanation: The loss of MHC Class I expression on tumour cells is a well-known mechanism of immune evasion, as MHC Class I molecules are essential for presenting tumour-specific antigens to CD8+ T-cells. Without this presentation, the immune system is unable to recognise and destroy the tumour cells. This allows the tumour to escape immune surveillance, even in the presence of immunotherapy. The loss of MHC Class I is a common immune escape mechanism seen in various cancers, including melanoma, and contributes to resistance to immunotherapy.
Option A is incorrect because although CD8+ T-cells may be recruited, their inactivation by CTLA-4 signalling from Tregs would not explain the loss of antigen recognition due to impaired MHC Class I expression.
Option C is incorrect because the upregulation of PD-1 receptors on tumour cells is typically associated with immune evasion by blocking T-cell activation, but the loss of MHC Class I expression directly impairs antigen presentation, which is the main issue in this case.
Option D is incorrect because although MDSCs (myeloid-derived suppressor cells) can inhibit T-cell infiltration, this mechanism is less relevant when the primary issue is the loss of MHC Class I expression, which directly prevents the recognition of tumour antigens.
Option E is incorrect because while the tumour may induce a fibrotic ECM to block T-cell infiltration, this is not the primary mechanism of immune evasion in this case. The loss of MHC Class I expression is the main cause of immune escape.
