

Paediatrics and genetics Answers (Y2)
Question 1:
Answer: C) Weaning may begin around 6 months, even if the infant is formula-fed
Explanation: WHO and UK guidelines recommend exclusive breastfeeding or formula-feeding for the first 6 months, followed by introduction of complementary foods. This applies regardless of feeding method.
A) is incorrect because delaying weaning to 12 months is not recommended as it can lead to deficiencies.
B) is incorrect as no solids should be given before 17 weeks due to immature gut.
D) is incorrect because early weaning has not been shown to prevent celiac and increases the risk of allergies.
E) is incorrect as all infants need weaning by ~6 months regardless of milk source.
Question 2:
Answer: E) Suggests acute malnutrition or feeding problem
Explanation: A drop across centiles in weight while length and head circumference are maintained suggests acute undernutrition or an issue with intake or absorption.
A) is incorrect because genetic short stature affects length, not weight alone.
B) is incorrect because although weaning can be a transition period, but significant centile drops are not normal.
C) Is incorrect because premature growth is corrected on charts only up to 36 months and would involve all parameters, the child would also show consistent prematurity.
D) is incorrect because catch-down growth refers to height, not isolated weight.
Question 3:
Answer: B) Plot weight 12 weeks earlier to account for corrected gestational age
Explanation: When plotting growth of a premature baby (<37 weeks), correct for gestational age until the child is 36 months old. So, subtract 12 weeks from actual age.
A) is incorrect because this would overestimate size.
C) is incorrect as correction is necessary up to 36 months.
D) is incorrect as there is no separate chart up to age 12; use UK-WHO with correction.
E) is incorrect as you must monitor growth from birth; you don’t delay plotting.
Question 4:
Answer: D) It provides complete nutrition and immune protection
Explanation: Breast milk is the gold standard for complete nutrition, especially with its immunological benefits such as passing maternal antibodies to the child therefore reducing the risk of infection, baby is less likely to get necrotising colitis, it protects the baby from diseases such as diabetes and CVD. It is also the perfect composition for infants and reduces the risk of the mother herself having breast cancer.
A) is incorrect because formula-fed infants may gain weight faster, but this is not ideal.
B) is incorrect since genetic conditions are unaffected by feeding method.
C) is incorrect as breastfeeding is not shown to delay teething significantly.
E) is incorrect as most infants tolerate formula lactose; true lactose intolerance in infancy is rare.
Question 5:
Answer: D) Child is overweight based on age- and sex-specific BMI chart
Explanation: Childhood BMI charts are age- and sex-specific. A BMI on the 98th centile suggests the child is overweight or obese for age. A BMI above the 91st percentile suggests overweight, and above the 98th percentile indicates very overweight or clinically obese, while a BMI below the 2nd percentile may suggest undernutrition.
A) is incorrect because the child’s height is on 75th centile, so there is no stunting.
B) is incorrect because there is no evidence BMI affects final height here.
C) is incorrect because adult BMI cutoffs do not apply to children.
E) is incorrect as because the height and weight centiles are not proportionally matched; the BMI confirms that this child is overweight.
Question 6:
Answer: B) Smooth pureed food
Explanation: Smooth purees are appropriate from 6 months.
A) is incorrect because chunkier foods are introduced later, around 10 months.
C) is incorrect because chopped adult meals it too advanced for initial weaning.
D) is incorrect as cow’s milk shouldn’t replace breast/formula until after 12 months.
E) is incorrect because raw proteins are unsafe in infancy.
Question 7:
Answer: A) Increased allergy and infection risk
Explanation: The gut and kidneys are immature before 6 months, and early food introduction increases risk of infection and allergies.
B) is incorrect because fat absorption is not directly impaired.
C) is incorrect as there is no direct evidence to suggest early weaning affects neurodevelopment.
D) is incorrect as hypercalcaemia is not a complication.
E) is incorrect as there is no evidence to support weaning improving sleep.
Question 8:
Answer: E) Tracheoesophageal fistula with oesophageal atresia
Explanation: TEF with oesophageal atresia causes NG tube coiling and aspiration with feeds.
A) is incorrect because laryngomalacia causes stridor, not aspiration.
B) is incorrect because a pyloric stenosis causes vomiting, not choking.
C) is incorrect because choanal atresia causes cyanosis that is relieved by crying.
D) is incorrect as laryngeal cleft is rare and presents similarly but there is no coiling.
Question 9:
Answer: C) Congenital diaphragmatic hernia
Explanation: Congenital diaphragmatic hernias cause bowel herniation into thorax resulting in lung compression.
A) is incorrect because tracheomalacia causes stridor.
B) is incorrect because BPD aka chronic lung disease is seen in preterm infants post-ventilation.
D) is incorrect because a TEF doesn’t explain scaphoid abdomen.
E) is incorrect because MAS is associated with meconium-stained amniotic fluid.
Question 10:
Answer: C) Trisomy 21
Explanation: Duodenal atresia is strongly associated with Down syndrome (Trisomy 21).
A) is incorrect because trisomy 13/ Patau’s disease is associated with cleft lip and holoprosencephaly.
B) is incorrect because trisomy 18/Edward’s syndrome results in clenched fists and rocker bottom feet.
D) is incorrect as monosomy X is Turner syndrome.
E) is incorrect because triploidy is typically nonviable.
Question 11:
Answer: E) (Mother’s height + 13 cm + Father’s height) / 2
Explanation: The estimated adult height of a child is calculated based on parental height using the following formulas based on the sec of the child: [(Mother’s height + 13 cm) + Father’s height] / 2 for a boy and[Mother’s height +( Father’s height – 13 cm)] / 2 for a girl.
A) is incorrect as it does not adjust for sex differences.
B) is incorrect because it sisapplies the ±13 cm adjustment.
C and D) are incorrect as this the formula for calculating a girl’s height.
Question 12:
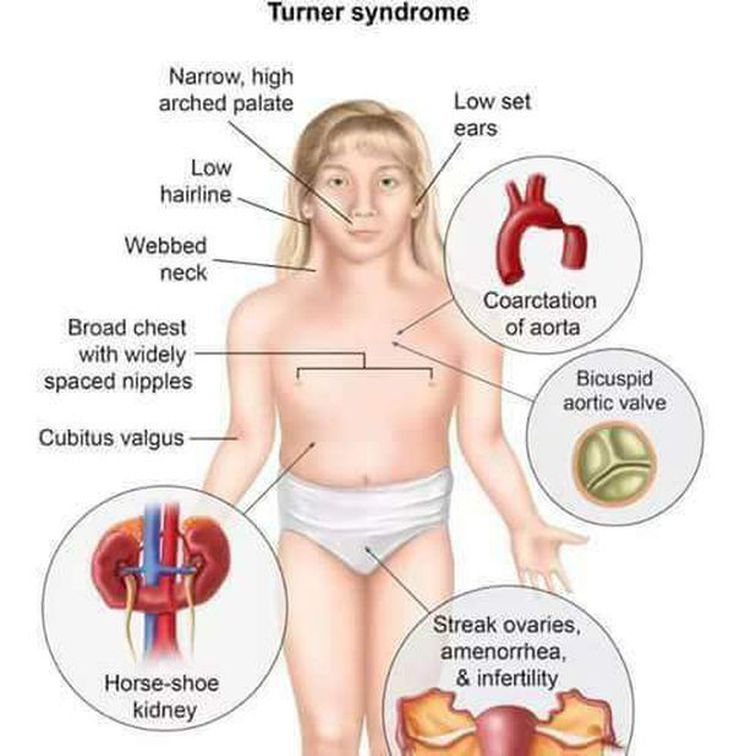
Answer: B) Widely spaced nipples and webbed neck
Explanation: Turner syndrome is a chromosomal abnormality with karyotype 45, XO. Patients are phenotypically female and have short stature, webbed neck, widely spaced nipples, and gonadal dysgenesis. Turner syndrome is associated with many additional abnormalities, including infertility, coarctation of the aorta, horseshoe kidney, and osteoporosis.
A) is incorrect because macrocephaly is not a feature of Turner syndrome.
D) is incorrect because Turner syndrome causes short stature and hypogonadism, it does not accelerate growth.
E) is incorrect as Turner’s causes delayed puberty not early puberty.
Question 13:
Answer: A) Growth hormone (GH)
Explanation: GH, in synergy with sex steroids, drives the pubertal growth spurt.
B) is incorrect because thyroxine supports general growth and is more important earlier in childhood but is not specific to puberty.
C, D and E) are incorrect as they are not directly involved in pubertal growth.
Question 14:
Answer: D) Growth hormone deficiency
Explanation: Normally, SGA babies catch up in size by 2 years old — meaning their growth curve improves to match other kids. If this does not happen it means, there is a problem with the normal growth process. Growth hormone (GH) is essential for normal growth after birth. It is one of the most important causes when SGA babies fail to grow. Usually these children have short stature, normal body proportions and delayed bone age on X-ray.
A) is incorrect because familial short stature means the child is small because the parents are small. But these children are born normal size and NOT SGA. They grow steadily but just stay short. However, this child was small at birth and failed to catch up which is not normal familial short stature.
B) is incorrect because Marfan Syndrome is a genetic disorder that causes a tall and thin build. A Marfan child would be taller on average than other children, not shorter.
C) is incorrect because hyperthyroidism causes children to grow fast initially not a failure to grow.
E) is incorrect because precocious puberty means early puberty and children with precocious puberty would initially grow quickly not slowly and be taller than expected not shorter.
Question 15:
Answer: B) Chromosome Microarray Analysis (CMA)
Explanation: Chromosome Microarray Analysis (CMA) is the first-line test for children with multiple congenital anomalies or developmental delays. It detects microdeletions and duplications such as the 22q11 deletion in DiGeorge syndrome.
A) is incorrect because karyotyping has lower resolution and misses small deletions. It also takes longer.
C) is incorrect as this is the test for Sickle cell anaemia
D) is incorrect as this is the test for cystic fibrosis specifically.
E) is incorrect because this tests specifically for Down syndrome
Question 16:
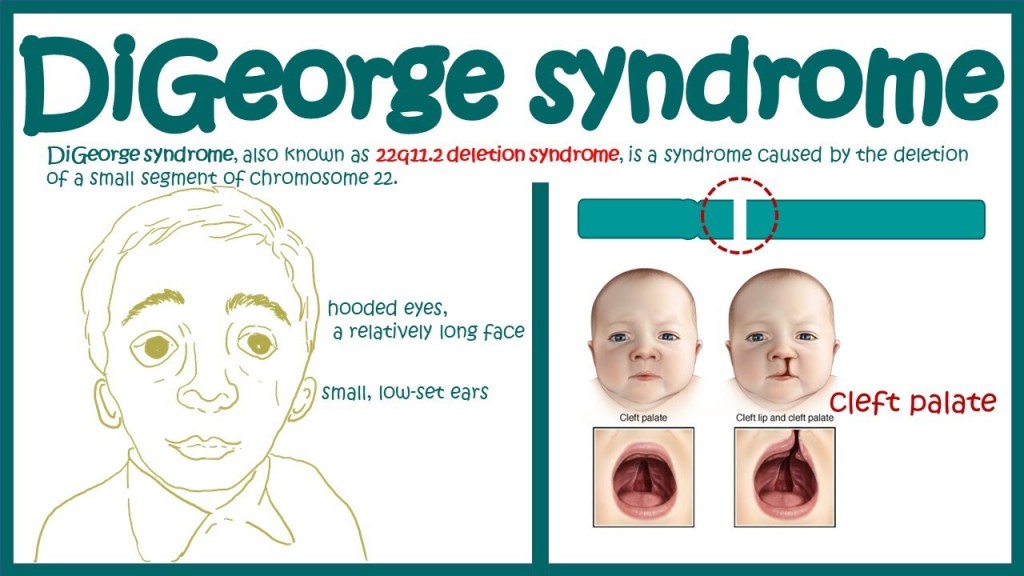
Answer: E) DiGeorge Syndrome (22q11 Deletion)
Explanation: DiGeorge syndrome is a congenital disorder caused by microdeletion on the long arm of chromosome 22. The clinical presentation includes features such as congenital heart defects such as TOF which causes cyanosis, craniofacial abnormalities, and/or defective development of the third and fourth pharyngeal pouches, resulting in hypoplastic thymus and parathyroids causing hypocalcaemia. CATCH-22 stands for Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate and Hypocalcaemia.
A) is incorrect because Down Syndrome includes intellectual disability and distinct facial features but not hypocalcaemia.
B) is incorrect because VACTERL are a group of associated birth defects consisting of Vertebral, Anal, and Cardiac abnormalities; Tracheoesophageal fistula; and Renal and Limb abnormalities.
C) is incorrect because Turnery syndrome causes infertility, coarctation of the aorta, horseshoe kidney, and osteoporosis.
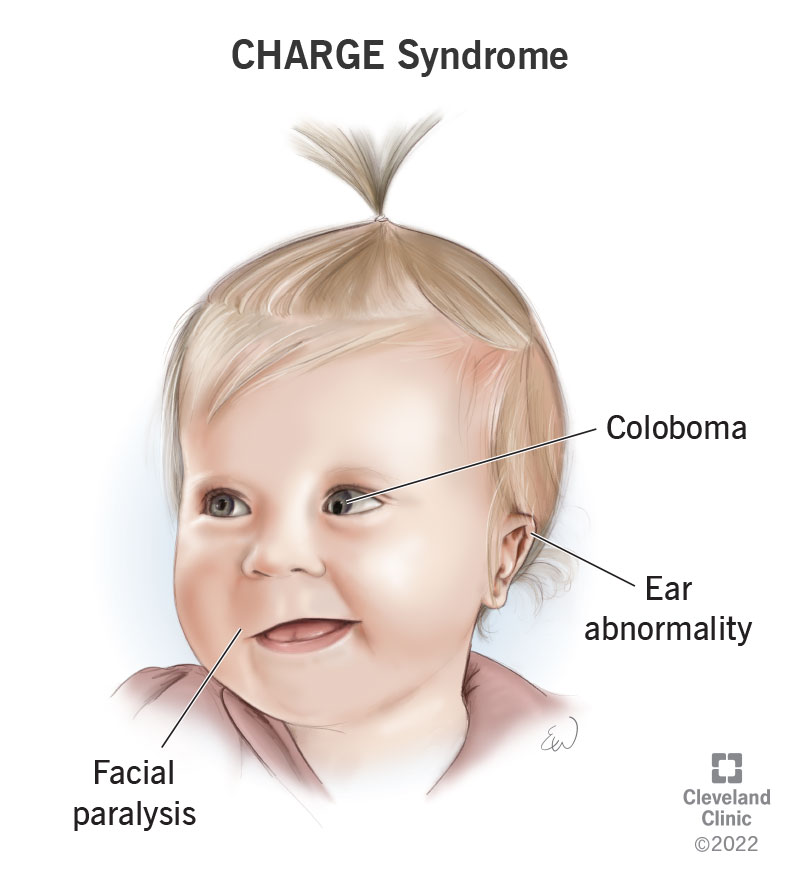
D) is incorrect because CHARGE is a syndrome of congenital anomalies consisting of Coloboma, Heart defect, Atresia of the choanae, Retardation of growth, Genital abnormalities, and Ear anomalies.

Question 17:
Answer: D) Pulmonary Hypoplasia
Explanation: Potter sequence is collection of foetal abnormalities caused by oligohydramnios – decreased amniotic fluid in pregnancy. The classical triad of Potter sequence is craniofacial abnormalities, clubbed feet, bilateral renal agenesis, and pulmonary hypoplasia – underdeveloped lungs impairing gas exchange.
A and B) are incorrect as these are symptoms of CHARGE SYNDROME.
C) is incorrect as thymic aplasia is a characteristic of DiGeorge syndrome
E) is incorrect as macrocephaly can be seen in many conditions such as acromegaly, hydrocephalus and Tay-Sachs disease etc.
Question 18:
Answer: B) Targeted BRCA1/2 Panel
Explanation: These genes are tumour suppressor genes located on the long arm of chromosome 17 and chromosome 13 respectively. Mutations in these genes can predispose to the development of breast and ovarian cancer. Targeted panels are performed when the genes for certain diseases on conditions in known.
A/C/D/E: Are incorrect because they are broader tests and less efficient for confirmed hereditary cancer genes.
Question 19:
Answer: C) Class 3
Explanation: VUS is Class 3 (uncertain significance) per ACMG guidelines it is a genetic change identified through testing, but the impact on health or function is not yet clear.
A and B) are incorrect as these are Class 1 and 2 respectively and are likely pathogenic.
D and E) are incorrect because Class 4 and 5 respectively are likely benign.
Question 20:
Answer: A) Whole Genome Sequencing (WGS) trio (proband + parents)
Explanation: VACTERL association is often sporadic with no single genetic cause. WGS trio can detect de novo variants or cryptic structural changes.
B) is incorrect because CHD7 is tested for CHARGE syndrome (coloboma, choanal atresia).
C) is incorrect because FISH uses a fluorescent DNA probe that sticks to a specific chromosome region, in TEF we do not know the specific chromosome region.
D) Mitochondrial disorders present with metabolic/neurologic symptoms.
E) BRCA1/2 relates to breast/ovarian cancer.
Question 21:
Answer: B) Collagen
Explanation: Osteogenesis imperfecta is a genetic disorder characterized by defective synthesis of type 1 collagen, which is important in bone formation. Patients present with signs that are sometimes mistaken for child abuse (e.g., easy bruising, predisposition to bony fractures). Additional clinical features include blue sclerae, joint laxity, hearing loss, and brittle, opalescent teeth.
A) is incorrect because Fibrillin-1 mutations cause Marfan syndrome.
C) is incorrect because dystrophin defects lead to muscular dystrophy.
D) is incorrect because keratin issues relate to skin disorders.
E) is incorrect because elastin leads to vascular disorders.
Question 22:
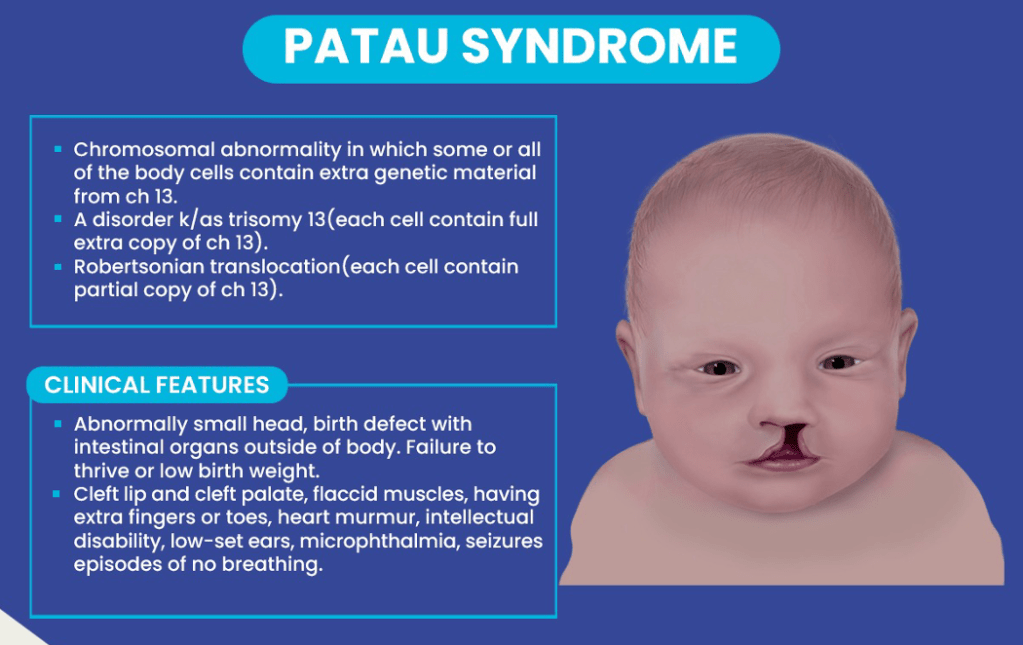
Answer: C) Patau syndrome
Explanation: Patau syndrome is a disease caused by trisomy of chromosome 13. It is the least common and most severe chromosomal trisomy. The most characteristic clinical features include microcephaly, cleft lip and palate, holoprosencephaly, polydactyly, and congenital heart defects.
A) is incorrect because Down syndrome has very different presentations
B) is incorrect because Edward syndrome has organ malformations congenital heart defects, horseshoe kidneys but not polydactyl.
D) is incorrect because Turner syndrome has coarctation of the aorta, horseshoe kidney, and osteoporosis.
E) is incorrect because DiGeorge syndrome causes congenital heart defects, craniofacial abnormalities, and/or defective development of the third and fourth pharyngeal pouches, resulting in hypoplastic thymus and parathyroids and not polydactyl.
Question 23:
Answer: B) Malrotation with volvulus
Explanation: Bilious vomiting (green vomit) in a neonate suggests intestinal obstruction beyond the duodenum, always assume this is malrotation with volvulus until proven otherwise as this is a surgical emergency.
A) is incorrect because meconium aspiration is a lung problem, not gut.
C) is partially correct because cystic fibrosis although it causes meconium ileus (gut obstruction), the first sign is not bilious vomiting at 6 hours old.
D) is incorrect because a tracheoesophageal fistula causes a feeding/choking problem, not bile vomiting due to an abnormal connection between the trachea and oesophagus.
E) is incorrect because a patent ductus arteriosus is a heart problem, not gut it is caused by the incomplete closure of the ductus arteriosus postnatally, resulting in a left-to-right shunt that causes a loud, continuous murmur.
Question 24:
Answer: E) Hernia on right side
Explanation: Right-sided diaphragmatic hernias are worse because they often pull the liver into the chest, severely affecting lung development (pulmonary hypoplasia). Left-sided hernias are more common but have better outcomes.
C) is incorrect because no liver involvement means better prognosis
B) is incorrect because this is not a prognostic feature involved
D) is incorrect because polyhydramnios does not worsen the prognosis even if present.
Question 25:
Answer: C) Edwards syndrome
Explanation: Edward syndrome is a congenital disease in which there is a third copy of chromosome 18 (trisomy 18). It is characterised by prominent occiput, low-set ears, clenched fists with flexion contractures of the fingers, rocker bottom feet, and organ malformations (congenital heart defects, horseshoe kidneys). It is the second most common trisomy in liveborn children.

Question 26:
Answer: A change in a single DNA base
Explanation: SNPs are a DNA sequence variant in a population that differ by only a single base pair, usually caused by errors during DNA replication and are point mutations. They are common and usually benign but can be associated with disease risk.

Question 27:
Answer: B) Fine motor
Explanation: Batting at objects reflects fine motor development, specifically voluntary hand-eye coordination which batting involves. At 4 months, infants use a palmar grasp but cannot yet transfer objects.
A) is incorrect because a gross motor at 4 months includes head control and arm pushing during tummy time.
C) is incorrect because language milestones at this age include turning to sound.
D) is incorrect because social milestones include interactive smiling more interactively and laughing
Question 28:
Answer: A) Moro reflex.
Explanation: The Moro reflex typically disappears by 4-6 months. Persistence beyond 6 months may indicate CNS dysfunction (e.g., cerebral palsy).
B) is incorrect as the parachute reflex appears at 6-8 months and persists forever.
C) is incorrect because the Babinski reflex (toe fanning) is normal in infants but abnormal after age 2.
D) is incorrect because the licking reflex does not exist.
E) is incorrect because trunk incurvation disappears by 6-9 months.

Question 29:
Answer: C) Exclusive breastfeeding significantly reduces the incidence of necrotising enterocolitis (NEC) in neonates, particularly in preterm infants.
Explanation:
Breast milk is protective against NEC, a serious and life-threatening intestinal disease primarily affecting premature babies. Breast milk contains immune factors (e.g., secretory IgA, lactoferrin, growth factors) that strengthen gut integrity and decrease inflammation, which dramatically lowers the risk of NEC compared to formula-fed infants.
- Incorrect: Breastfeeding reduces risk of both Type II Diabetes and cardiovascular disease, not just diabetes.
- Incorrect: IgA, not IgG, is the main antibody transferred via breast milk (IgG crosses via placenta).
- Incorrect: Although 74% of mothers start breastfeeding, only about 43% continue at 6–8 weeks in the UK — not over 70%.
- Incorrect: Any breastfeeding (even for a short time) lowers breast cancer risk; it’s not limited to breastfeeding beyond 2 years.
Question 30:
Answer: C) Delay weaning until around 6 months, when developmental milestones like head control and swallowing are achieved.
Explanation:
Weaning should start at around 6 months, when infants are developmentally ready (able to hold their head up and swallow safely). Introducing solids too early (before 6 months) increases the risk of allergies, infections, and places strain on immature gut and kidney systems.
- A) Incorrect: The gut and kidneys are not fully mature at 5 months; weaning before 6 months is not advised.
- B) Incorrect: Early introduction of solids increases allergy risk, rather than reducing it.
- D) Incorrect: Initial weaning (6–8 months) should start with smooth purees, not chopped-up adult food.
- E) Incorrect: Infants need higher fat intake during early weaning for proper growth and energy needs.
Question 31:
Answer: C) Numerical abnormality: trisomy
Explanation:
An extra copy of a single chromosome (like chromosome 21 in Down’s Syndrome) is called a trisomy, which is a numerical chromosomal abnormality affecting one chromosome pair.
- A) Incorrect: Reciprocal translocation involves exchange of sections between two chromosomes, not the addition of a full extra chromosome.
- B) Incorrect: Deletion refers to loss of a section of DNA, not gaining a full chromosome.
- D) Incorrect: Monosomy means loss of one chromosome, not a gain.
- E) Incorrect: Triploidy is when an entire extra set of chromosomes is present (69 chromosomes total), not just one extra chromosome.
Question 32:
Answer: B) Structural abnormality: Robertsonian unbalanced translocation involving an acrocentric chromosome
Explanation:
A Robertsonian translocation involves fusion of two acrocentric chromosomes (chromosomes 13, 14, 15, 21, or 22). In this case, material from chromosome 21 has moved onto chromosome 14. If the translocation is unbalanced, there is a gain of genetic material (extra chromosome 21 material), resulting in a Down’s Syndrome phenotype without classical trisomy.
- A) Incorrect: Classic trisomy 21 involves 47 chromosomes, not 46.
- C) Incorrect: A reciprocal translocation swaps material between two chromosomes but usually does not involve fusion or affect acrocentric chromosomes in this way.
- D) Incorrect: Triploidy involves a whole extra set of chromosomes (69 chromosomes total), not a specific rearrangement between two chromosomes.
- E) Incorrect: A deletion would involve loss of chromosomal material, not a gain of chromosome 21 material.
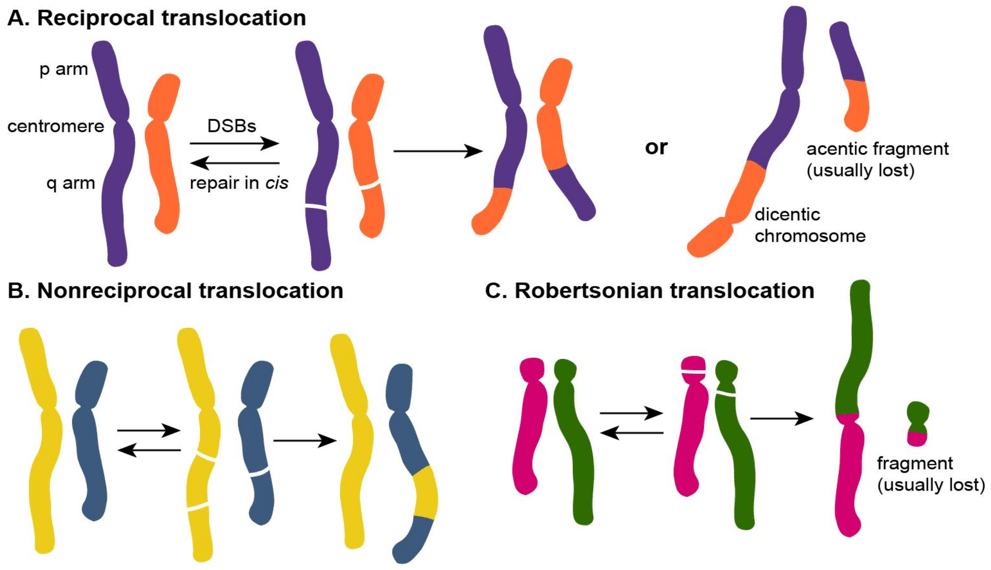
Question 33:
Answer: C) Patau Syndrome (Trisomy 13)
Explanation:
Patau Syndrome results from trisomy 13 and is characterized by severe congenital abnormalities such as cleft lip/palate, polydactyly, cardiac defects, and profound intellectual disability. It carries a very poor prognosis.
- A) Incorrect: Edwards Syndrome (Trisomy 18) also causes congenital defects, but typically presents with clenched fists with overlapping fingers and rocker-bottom feet, not polydactyly.
- B) Incorrect: Turner Syndrome (45, X) affects females but typically presents with short stature, webbed neck, and primary amenorrhea, not multiple congenital anomalies.
- D) Incorrect: Klinefelter Syndrome (47, XXY) affects males and presents with hypogonadism, infertility, and tall stature — not congenital physical anomalies at birth.
- E) Incorrect: Triple X Syndrome (47, XXX) usually has minimal or no physical abnormalities and often goes undiagnosed.
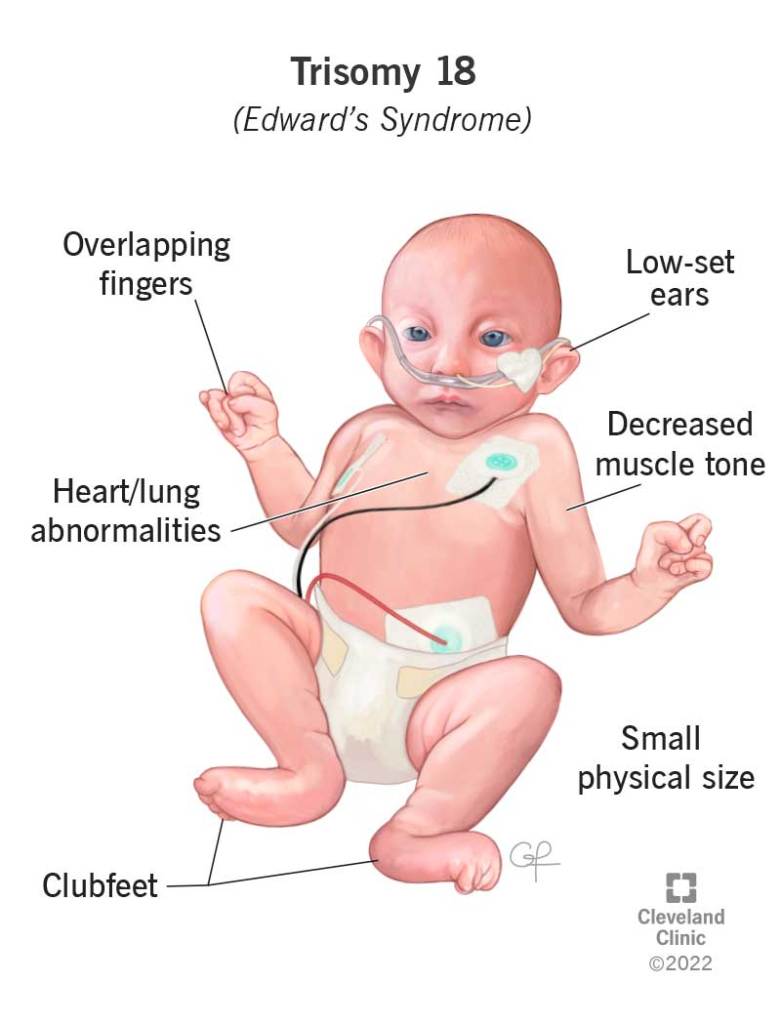

Question 34:
Answer: B) Multicoloured FISH + Microarray Analysis
Explanation:
Multicoloured FISH + Microarray Analysis allows for the detection of duplications and deletions at a higher resolution than traditional methods. This technique uses fluorescent probes in different colours to identify specific chromosomal abnormalities and can reveal gains or losses of chromosomal material by comparing the patient’s sample to a control.
- A) Incorrect: FISH with a probe for chromosome 21 would specifically detect trisomy 21 or other structural issues related to chromosome 21, but would not detect small-scale duplications or deletions across the genome.
- C) Incorrect: Next Generation Sequencing (NGS) can identify single nucleotide polymorphisms (SNPs) and small sequence variants, but it is not the most efficient method for detecting large-scale chromosomal duplications or deletions.
- D) Incorrect: A Genome-Wide Association Study (GWAS) examines genetic variation across a large population to identify associations between genes and traits, but it does not specifically target duplications or deletions in a single patient.
- E) Incorrect: Karyotyping can detect large-scale chromosomal abnormalities, such as trisomies or large deletions, but it has low resolution for detecting small duplications or deletions.
Question 35:
Answer: B) The entire amino acid sequence after the mutation will be altered, and the protein may be non-functional.
Explanation:
The insertion of a single base pair into a gene causes a frameshift mutation. Since each codon consists of 3 nucleotides, the insertion shifts the reading frame of the gene, altering the amino acid sequence downstream of the mutation. This will result in a completely different sequence of amino acids and likely a non-functional protein due to the disruption of its structure and function.
- A) Incorrect: While a premature stop codon can occur in some mutations, a single base insertionwill primarily cause a frameshift, which alters the amino acid sequence, not necessarily a premature stop.
- C) Incorrect: A silent mutation would not cause any change in the amino acid sequence, which is not the case here since frameshift mutations result in a different amino acid sequence.
- D) Incorrect: The silent mutation typically does not change the protein, but a frameshift mutation will change the sequence of amino acids.
- E) Incorrect: Insertion of a single base pair will alter the reading frame, leading to major effects, not no effect.
Question 36:
Answer: B) The next daughter will have a 50% chance of being a carrier of DMD.
Explanation:
For Duchenne’s Muscular Dystrophy (DMD), which is an X-linked recessive disorder, the woman is a carrier, meaning she has one affected X chromosome and one normal X chromosome. Her partner is unaffected and thus has a normal X and Y chromosome. Sons inherit their X chromosome from their mother and a Y chromosome from their father. Since the mother is a carrier, each son has a 50% chanceof inheriting the affected X chromosome, which would result in the son being affected by DMD (because males are hemizygous for the X chromosome). Daughters inherit one X chromosome from each parent. Since the father is unaffected and provides a normal X chromosome, each daughter has a 50% chance of inheriting the affected X chromosome from her mother. If she does, she will be a carrier, but she will not express DMD because the second, normal X chromosome compensates. However, X-inactivation /lionisation of an X chromosome in females can occur. If the wild-type X chromosome is inactivated, then the daughter will have the disease, but the question did not hint at lionisation thus it is safe to assume that the daughter will be a carrier.
- A) Incorrect: Sons cannot inherit a second X chromosome from their father (they inherit a Y chromosome), so there is no possibility of a 25% chance. Sons have a 50% chance of being affected if they inherit the affected X chromosome from the mother.
- C) Incorrect: Sons cannot be carriers of X-linked disorders because males only have one X chromosome. If they inherit the affected X, they will be affected.
- D) Incorrect: Daughters cannot be affected by DMD unless they inherit two affected X chromosomes, which is not possible here, as the father contributes a normal X. Therefore, the daughter’s risk of being affected is 0%.
- E) Incorrect: The risk for sons is 50% for being affected, not 50% for being a carrier, as only femalescan be carriers. Sons cannot be carriers of X-linked disorders.
Question 37:
Answer: D) The loss of paternal gene expression occurs due to methylation or deletion of a specific region on chromosome 15, and since the maternal allele is silenced, the child has no functional copy of the gene.
Explanation:
Prader-Willi syndrome (PWS) is caused by the loss of the paternal contribution to a specific region on chromosome 15. The affected gene region is typically silenced on the maternal allele due to imprinting, and deletion or methylation of the paternal allele leads to the loss of functional gene expression. This loss of function in the affected region results in the characteristic features of Prader-Willi syndrome, such as hypotonia, developmental delays, and insatiable hunger.
- A) Incorrect: In PWS, the child inherits one normal copy of chromosome 15 from the mother and one affected copy from the father, not two copies from the mother. The lack of paternal gene expression due to methylation or deletion causes the syndrome.
- B) Incorrect: In PWS, it is the loss of the paternal contribution (not the maternal allele) that causes the disease. The maternal allele is imprinted (silenced) in the region of interest.
- C) Incorrect: The failure of X-inactivation does not contribute to Prader-Willi syndrome. The issue in PWS is the loss of paternal gene expression due to either deletion or methylation, not X-inactivation.
- D) Correct: In PWS, deletion or methylation of the paternal allele results in the loss of function of the gene. Since the maternal allele is silenced due to imprinting, no functional copy of the gene is available, leading to the symptoms of the syndrome.
- E) Incorrect: In Prader-Willi syndrome, the paternal allele is not silenced by methylation but is lost or deleted. The maternal allele is the one that is silenced.
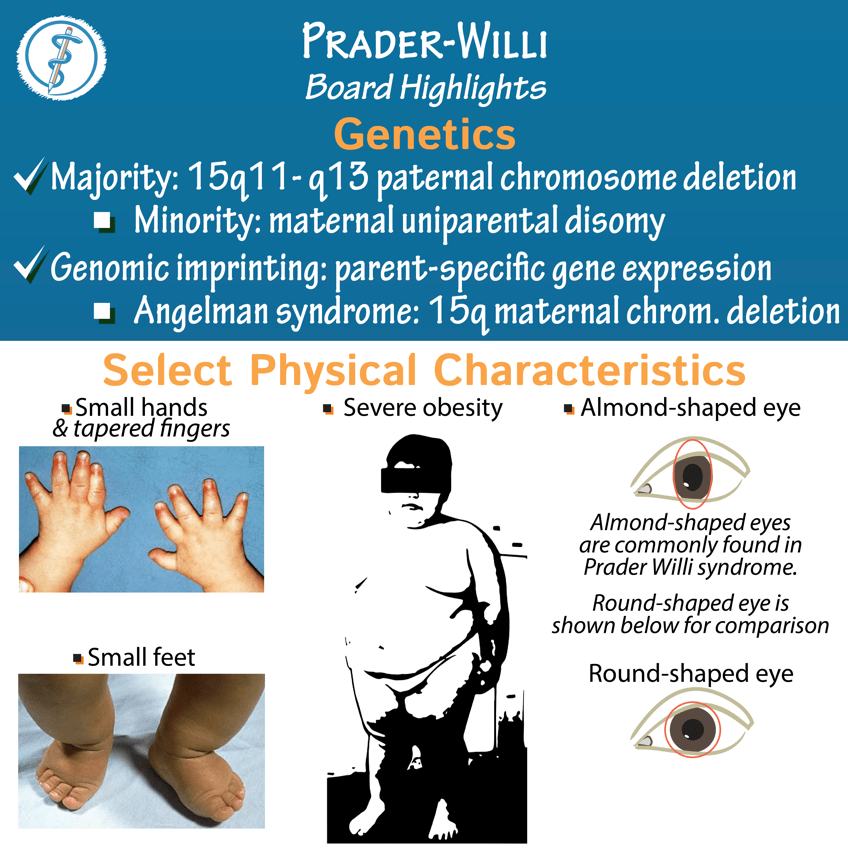

Question 38:
Answer: A) Trisomy 13 (Patau’s Syndrome), Trisomy 18 (Edwards Syndrome), VACTERL association, and Holt-Oram Syndrome
Explanation:
Tracheoesophageal fistula (TEF) is a congenital malformation that can be associated with a variety of syndromes and genetic conditions. The most common type of TEF is type C. The following are correctly associated with TEF:
- Trisomy 13 (Patau’s Syndrome): This genetic condition is associated with multiple congenital defects, including TEF, cleft lip/palate, and heart defects.
- Trisomy 18 (Edwards Syndrome): Like Trisomy 13, Edwards syndrome can involve TEF as part of the broader spectrum of congenital anomalies, including cardiac defects and growth restrictions.
- VACTERL association: This non-random group of birth defects, including vertebral, anal, cardiac, tracheoesophageal, renal, and limb defects, is known to be associated with TEF.
- Holt-Oram Syndrome: A genetic condition that causes heart defects and upper limb abnormalities. It is linked to TEF in some cases.
- B) Incorrect: Trisomy 21 (Down syndrome) is not typically associated with TEF. Although Cystic Fibrosis affects the lungs and digestive system, it does not typically cause TEF.
- C) Incorrect: DiGeorge Syndrome and Duchenne Muscular Dystrophy are not typically linked with TEF, though DiGeorge may cause heart defects.
- D) Correct: CHARGE Syndrome involves coloboma, heart defects, atresia of the choanae, retardation of growth, and genital/ear anomalies. It is also associated with TEF, so this is a viable alternative answer.
- E) Incorrect: Beckwith-Wiedemann Syndrome is typically not associated with TEF, though it is known for overgrowth and organ enlargement.
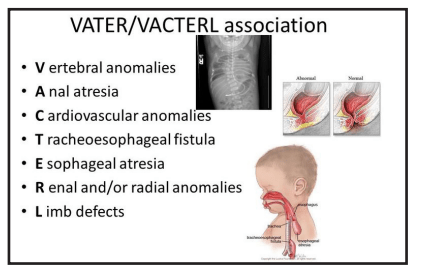
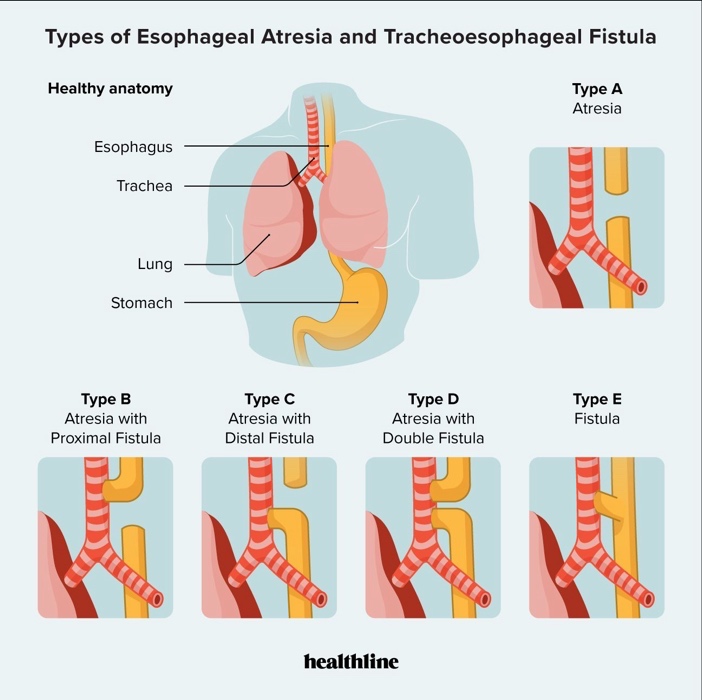
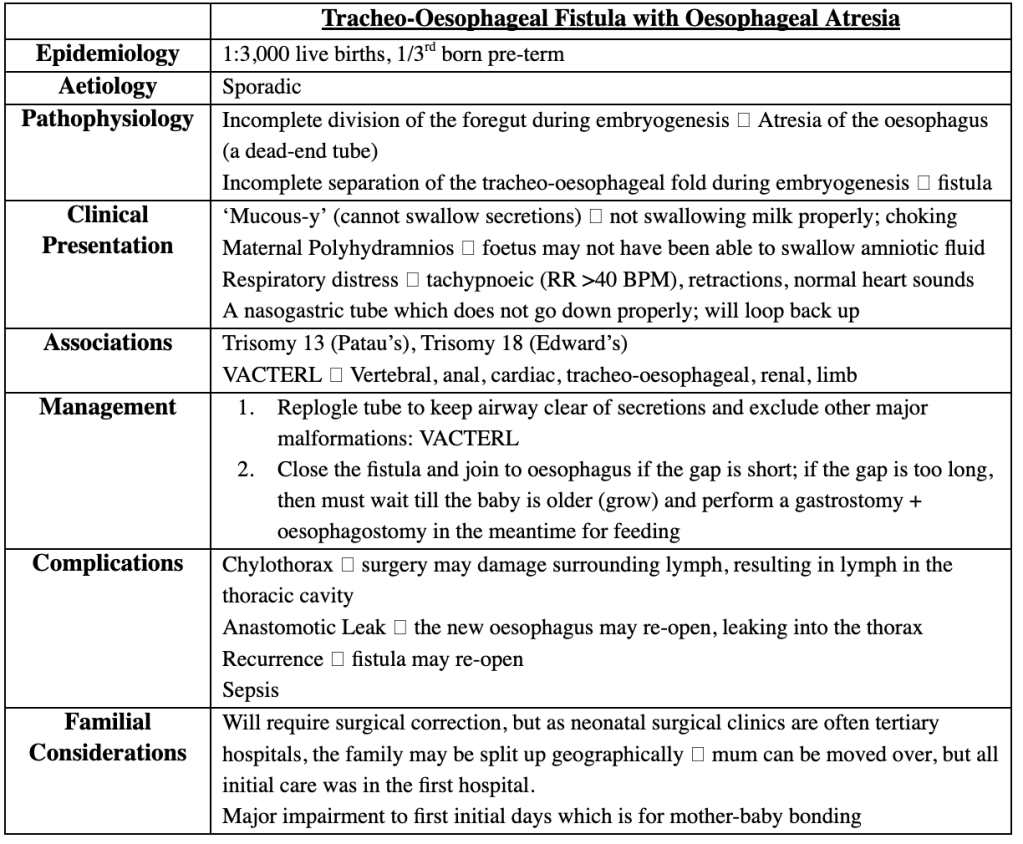
*Credits to Dr Aarifa Khanom for this table
Question 39:
Answer: B) Malrotation with volvulus; midgut ischemia due to superior mesenteric artery compromise
Explanation:
This neonate has classic signs of intestinal obstruction with bilious vomiting, abdominal distension, minimal stool output, and non-specific X-ray findings — all raising concern for a high small bowel obstruction.
The key diagnostic clue is the risk of SMA strangulation causing midgut ischemia, making malrotation with volvulus the most urgent consideration. Contrast upper GI series would be diagnostic (abnormal duodenojejunal position).
- A) Incorrect: Pyloric stenosis presents after 2-6 weeks of life, causes non-bilious, projectile vomiting, and metabolic alkalosis.
- C) Incorrect: Hirschsprung’s can cause bowel obstruction, but not early volvulus or SMA compromise.
- D) Incorrect: Duodenal atresia would show a “double bubble” sign on X-ray, not a non-specific gas pattern.
- E) Incorrect: Meconium ileus presents with distal obstruction signs and association with cystic fibrosis, not bilious vomiting with proximal signs.
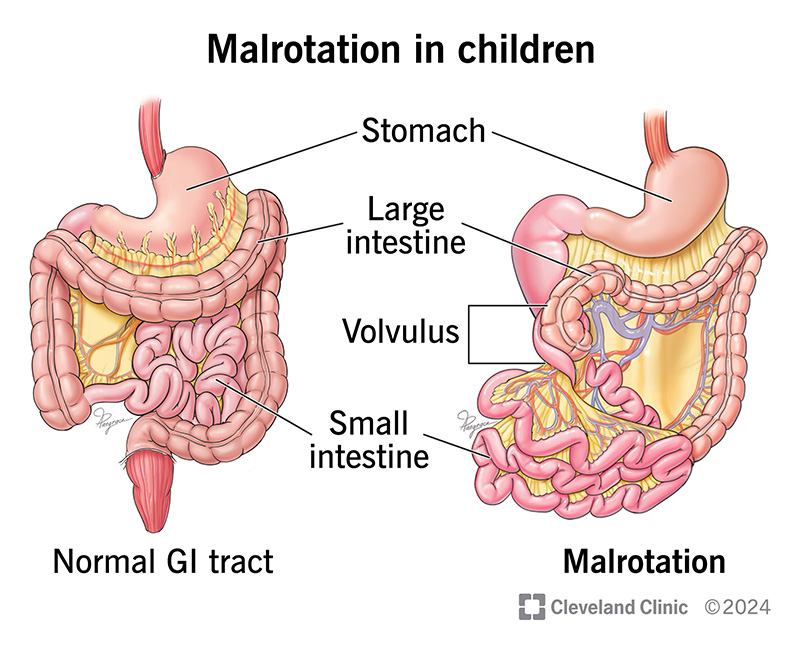
Question 40:
Answer: C) Pulmonary valve stenosis
Explanation:
This infant shows signs of right ventricular outflow obstruction (cyanosis after ductal closure, normal lungs, poor saturations, sternum pulsations).
Pulmonary valve stenosis leads to RV pressure overload, RV hypertrophy, and post-stenotic pulmonary trunk dilation.
Diagnosis is confirmed with echocardiography.
- A) Incorrect: Tetralogy of Fallot would show right-to-left shunting but often has a harsh systolic murmur and not isolated pulmonary trunk dilation.
- B) Incorrect: PPHN would show respiratory distress and elevated pulmonary pressures without structural heart defects.
- D) Incorrect: TGA would present with profound cyanosis immediately after birth and needs mixing lesions (e.g., ASD, PDA) to survive.
- E) Incorrect: Coarctation affects systemic perfusion, leading to poor femoral pulses, differential cyanosis, and not primarily pulmonary signs.
Question 41:
Answer: A) Activity-dependent synaptic proliferation enhancing neural plasticity
Explanation:
At 2 years old, the brain is in a critical window of hyperactive synaptogenesis driven by external experience.
- Birth to 3 months: Rapid formation of synapses begins (~1,000 trillion synapses).
- By 3 years: About 80% of synapses are already formed, and the brain is hungrily responding to external stimuli.
- This activity-dependent plasticity allows rapid cognitive, emotional, and motor development.
- Pruning of unused synapses does not dominate until after 11 years, meaning that synaptic growth — not elimination — is the main feature at age 2.
- Nutritional needs are especially high in this period (up to 60% of nutritional intake directed to brain activity).
- B) Incorrect: Pruning does occur but later (after ~11 years) and is not the dominant process at this young age.
- C) Incorrect: Programmed apoptosis mainly regulates neuronal numbers prenatally, not synaptic changes postnatally.
- D) Incorrect: Myelination is ongoing but not the primary plastic event during explosive cognitive development.
- E) Incorrect: Early brain circuits are not fully stabilised; they are dynamic and moulded by environment and experience.
Question 42:
Answer: B) Early sign of upper motor neuron dysfunction
Explanation:
By 4–6 months, infants should demonstrate:
- Good head control, ability to press up/pull up while prone
- Grasping and transferring objects between hands
- Social responses like recognising their own name
Reflexes such as the asymmetric tonic neck reflex (ATNR), Moro, palmar grasp, and rooting reflex are normally present at birth but should disappear by 4–6 months.
Persistence of primitive reflexes beyond 6 months suggests underlying neurological impairment, most often upper motor neuron dysfunction (e.g., cerebral palsy).
- A) Incorrect: Persistence of primitive reflexes at 5 months is not normal; it raises concerns for CNS dysfunction.
- C) Incorrect: Fine motor skills are progressing (grasping, transferring objects); it’s the primitive reflex persistence that signals abnormality.
- D) Incorrect: Cerebellar hypoplasia primarily affects balance and coordination later in infancy/toddlerhood, not early reflex disappearance.
- E) Incorrect: Reflex persistence is pathological after 4–6 months; it’s not a benign variant.
Question 43:
Answer: A) Normal for age
Explanation:
At 2 years of age, the expected developmental milestones include:
- Gross Motor: Walks steadily, climbs stairs using two feet per step.
- Fine Motor: Tripod grip starting, basic scribbling.
- Language: Combines 2–3 words together (“more juice”)
- Social: Begins undressing, though may still need help
Thus, everything described (walking with two feet per step, scribbling, two-word phrases, partial undressing) fits normal developmental expectations for a 2-year-old.
- B) Incorrect: Two feet per step at stairs is normal for age; gross motor is appropriate.
- C) Incorrect: Scribbling is expected fine motor behaviour at 2 years; no delay.
- D) Incorrect: Two-word phrases are expected at 2 years; language is appropriate.
- E) Incorrect: Partially undressing is normal at 2 years; no social delay.
Question 44:
Answer: B) 3 years
Explanation: At this stage, the typical developmental milestones are:
- Gross Motor: The ability to climb stairs with one foot per step and the emergence of hopping with support.
- Fine Motor: The ability to copy a circle, demonstrating improvement in drawing skills.
- Language: The use of short sentences (3-4 words) and basic pronouns like “me,” “I,” and “you.”
- Social: The ability to name friends and becoming more independent with self-care tasks like being out of nappies.
Why the other options are incorrect:
- A) 2 years: Children at this stage typically undress, begin to use 2-word combinations, and show basic social behaviours, but they aren’t using complex 3-word sentences or drawing more refined shapes like a circle.
- C) 4 years: At 4 years, children start to copy a triangle, use more complex sentences, and show greater social maturity in play.
- D) 5 years: At 5 years, children skip, draw more complex shapes, and develop more advanced language and social play skills.
- E) 6 years: At 6 years, children have refined social play, clearer speech, and more advanced fine motor skills, such as writing and drawing with more detail.
Question 45:
Answer: B) Gross motor: Lifting head while prone, but unable to support head for long
Explanation:
At 6-8 weeks, the typical developmental milestones for an infant would be:
- A) Fine motor: At this age, infants are not yet reaching for or grasping objects with both hands; this typically begins around 3-4 months as they develop fine motor coordination.
- B) Gross motor: Infants at 6-8 weeks can lift their head briefly while prone but cannot support it for extended periods. They are developing neck strength and beginning motor control, which is a key developmental milestone in this stage.
- C) Social: While infants may start showing social smiles, they are not typically distinguishing between familiar faces and strangers yet. This ability develops around 3-4 months.
- D) Speech: Infants at 6-8 weeks may start vocalising sounds, mainly throat sounds, but they are not responding to speech or producing recognisable words at this point.
- E) Cognitive: Infants are indeed beginning to track objects briefly with their eyes, but this would be a more accurate description of a 2-3 month old, when visual tracking becomes more refined. At 6-8 weeks, they may fixate on objects but not track them continuously.
Question 46:
Answer: B) CMA can identify submicroscopic chromosomal deletions and duplications, which may not be detectable by conventional karyotyping, thus helping explain the child’s developmental delay.
Explanation:
In this case, the child presents with developmental delay and dysmorphic features, suggesting a possible chromosomal abnormality. However, the standard karyotype (which detects large chromosomal abnormalities like aneuploidies) fails to identify any issues. This is where chromosome microarray testing (CMA) proves particularly beneficial.
- A) Incorrect: CMA does not primarily focus on single nucleotide polymorphisms (SNPs), which are typically associated with point mutations. These require different testing techniques, such as next-generation sequencing (NGS), not CMA.
- B) Correct: CMA can identify submicroscopic chromosomal deletions and duplications (often referred to as copy number variations (CNVs)) that may not be visible through conventional karyotyping. This is especially important in cases of developmental delay or dysmorphic featureswhere small chromosomal abnormalities could be the underlying cause.
- C) Incorrect: While CMA is useful for detecting small chromosomal deletions and duplications, it is not used for detecting large chromosomal aneuploidies like trisomy 21, which are best diagnosed using karyotyping or FISH.
- D) Incorrect: CMA is not designed for detecting epigenetic modifications such as DNA methylation. Specific tests like methylation-sensitive techniques are required for diagnosing conditions like Prader-Willi or Angelman syndrome.
- E) Incorrect: CMA does not detect mitochondrial DNA mutations. Mitochondrial disorders require specialised testing, such as mtDNA sequencing, and are not addressed by chromosome microarray technology.

Question 47:
Answer: A) Disruption of neural crest cell migration leading to abnormalities in the development of the third and fourth pharyngeal pouches.
Explanation:
DiGeorge syndrome (also known as 22q11.2 deletion syndrome) is caused by a microdeletion on chromosome 22, which affects several embryological processes and organ systems. The critical embryological defect lies in the development of neural crest cells, which are essential for the formation of several structures, particularly those arising from the third and fourth pharyngeal pouches.
- A) Correct: The third and fourth pharyngeal pouches give rise to critical structures, including the thymus and parathyroid glands, which are often hypoplastic or absent in DiGeorge syndrome. Neural crest cells, which migrate from the neural tube during early embryogenesis, play a key role in the development of these pouches. A failure in the migration or development of these cells leads to the characteristic immune deficiency (due to thymic hypoplasia), hypocalcaemia (due to parathyroid hypoplasia), and congenital heart defects seen in DiGeorge syndrome.
- B) Incorrect: While mesodermal differentiation is critical for proper cardiac development, the primary defect in DiGeorge syndrome involves the neural crest cells, not mesodermal differentiation.
- C) Incorrect: Mesonephric duct development affects kidney and ureteral formation, but is unrelated to DiGeorge syndrome, which is primarily characterised by abnormalities in neural crest cell migration and pharyngeal development.
- D) Incorrect: The ectodermal layer does give rise to neural structures, but the pathophysiology of DiGeorge syndrome involves the failure of neural crest cells, not ectodermal differentiation.
- E) Incorrect: Notochord development is crucial for axial skeletal formation, but DiGeorge syndromeis not caused by defects in the notochord, rather it involves neural crest cell migration.
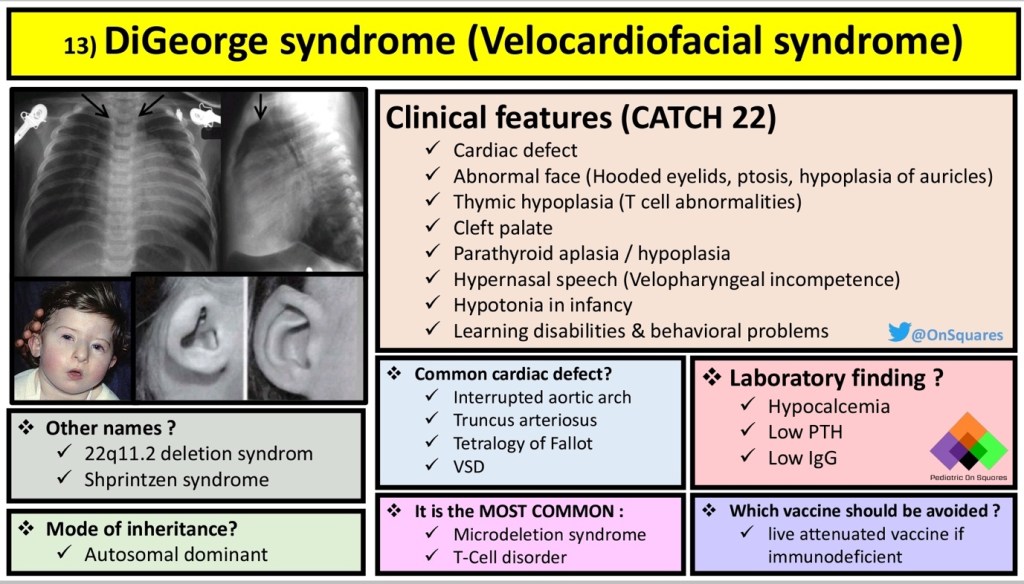
Question 48:
Answer: B) The variant alters a known functional domain of the BRCA1 protein, but its effect on protein function remains unclear due to inconsistent experimental findings.
Explanation:
Variants of Uncertain Significance (VUS) are genetic variants whose association with a particular condition is unclear. These variants do not have enough evidence to either confirm or exclude their role in causing disease. In the case of SNPs in critical genes like BRCA1, the classification as VUS indicates that the variant has been detected, but its functional impact on the gene or protein cannot be conclusively determined.
- A) Incorrect: While the SNP may increase the risk in some familial studies, a VUS classification implies that there is insufficient or inconsistent evidence to confirm the variant’s clinical significance. It may not be conclusively linked to breast cancer risk across broader populations.
- B) Correct: The variant alters a functional domain of the BRCA1 protein, which is crucial for DNA repair and tumour suppression. However, due to conflicting results from experimental studies, the exact effect of the variant on protein function remains uncertain. This is the typical scenario for a VUS classification: functional effects are suggested but not proven or consistent.
- C) Incorrect: A VUS is characterized by uncertainty. If the variant were benign, it would not be classified as a VUS; it would instead be classified as “benign” based on consistent evidence from cohort studies.
- D) Incorrect: Variants located in non-coding regions can affect regulatory elements, but without direct functional evidence supporting a role in cancer, they would not typically be classified as VUS unless there were experimental data indicating a possible functional effect, even if the mechanism is unclear.
- E) Incorrect: Variants in highly conserved regions may suggest functional importance, but for a VUS classification, there would still be insufficient evidence to suggest a definitive pathogenic role. A variant that doesn’t alter protein function is less likely to be pathogenic, making this answer unlikely.
